Introduction
MiRNAs (miRs) are small endogenous RNAs made up of 10 to 24 nucleotides via sequential processing of Iry RNAs responsible for inhibition of gene expression at the posttranscriptional level. The sequential processing of Iry transcripts is arbitrated by two enzymes, viz Dicer and Drosha. miRs have been found to play important roles in regulating cellular development, metabolism and immunity [1, 2]. miRs are also reported to be negative regulators of genes which inhibit the translation or promote the degradation of target RNAs [3]. In a recent study, mice reported to be cardiac-specific dicer deficient showed cardiac abnormalities such as heart failure, sudden cardiac death and dilated cardiomyopathy [4, 5]. Microarray analysis on human cardiac tissues and rats has revealed expression of miRs under various cardiac pathophysiology such as myocardial infarction, cardiac hypertrophy and cardiac failure [6–10]. An earlier study suggested involvement of specific miRs in the cardiac tissues of genetically modified mice, for example, mice deficient in miR-1-2 demonstrated cardiac arrhythmia and malformations from their birth [11]. miR-133a has been found to regulate proliferation of cardiomyocytes and suppress the smooth muscle gene expression in the cardiac muscles [12]. These studies suggest the potential role of miRs in cardiac pathophysiology and its development.
Contraction of cardiac muscles is dependent on expression levels of 2 cardiac myosin heavy chain genes (MHC) named α- and β-MHC. The expression of these two genes is regulated in an diametric manner via the pathological signals and the physiology [13]. It is also found that β-MHC is over-expressed in cardiac failure and hypertrophy; hence identifying miRs that can regulate the expression of β-MHC may lead to new pathways in treating such cardiac disorders.
In a recent study, miR-208a, which is a cardiac-specific miRNA, was found to play a vital role in regulation of the β-MHC gene in genetically modified mice showing over-expression of miR-208a [14]. Thus, these kinds of studies confirm the usefulness of genetically modified animal models in determining involvement of any specific miRNA. However, in order to establish direct effects of specific miRs, in vitro studies are still needed as varied hemodynamic and hormonal effects can alter the levels of β-MHC and blur the in vivo results [15].
Thyroid hormones have been found to play an important role in maintaining homeostasis of the cardiovascular system under conditions affecting heart rate, contractility, peripheral resistance and diastolic function via the genomic and non-genomic pathways [16]. These multiple actions of thyroid hormones are mainly executed via the thyroid receptors (TRs) [17]. Briefly, the expression of α-MHC is upregulated by T3 hormone, whereas the levels of β-MHC are suppressed [17]. In addition, the changes in thyroid hormone signaling are linked to cardiac failure and cardiac hypertrophy [16, 17].
In the current study, we evaluated the expression of the MHC gene in rats transduced with Dicer siRNA and 17 miRs, previously reported to be over-expressed in cardiac failure or hypertrophy [18]. The transduction was done in the neonatal rat ventricular myocytes (NRVMs) using transfecting vector. We demonstrated that suppression of Dicer, which is an important enzyme for biosynthesis of miRs, decreased the levels of miRs, leading to down-regulation of the α-MHC gene. We also reported that miR-29a was an important factor responsible for regulation of α-MHC through the thyroid receptor-β1 (TR-β1) in NRVMs, and additionally we found that the levels of miR-29a were increased in the process of differentiation of stem cells or in the hypertrophic cardiac muscles of animals associated with over-expression of the α-MHC gene.
Material and methods
Cell culture
For the study, one-day-old Sprague-Dawley rats were used; the neonatal rat ventricular myocytes were isolated from them as described earlier [19] and were used for the study. The myocytes were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with FBS (10%), penicillin (1%), streptomycin (1%) in a 96-well plate at room temperature in presence of CO2. The DNA was transduced at 48 h after seeding. The cell cultures were exposed to tri-iodothyronine.
For in vitro studies OLA derived embryonic stem cells ht7 (ES-ht7) (ATCC, USA) were used. The cells were subjected to DNA transduction; the cells were plated (marked as day ‘0’) on plates coated with gelatin and media containing lentivirus and incubated for 24 h. The effect of gene silencing in ES-ht7 cells was assessed by replacing the lentiviral vectors with phosphoglycerate kinase promoter (PGK). After this 3 × 104 cells/well were cultured into gelatin coated 6-well plates devoid of leukemia inhibitory factor, embryonic bodies and feeder cells for 7 days.
Animals and transverse aortic constriction (TAC) procedure
The animal studies were approved by the animal ethical committee of Xiangyang Central Hospital, Hubei China. All the protocols were in accordance with guidelines laid down by the animal review board of Hubei University of Arts and Science Xiangyang, Xiangyang, China. For the current study C57 black/6 Jacksonian mice were used and were subjected to TAC as reported earlier [20]. The fetal ICR mice were evaluated for vaginal plugs and after 14 days the heart tissue samples were obtained. The obtained samples were subjected to quantitative reverse transcription-PCR (qRT-PCR).
Construction of vectors
The miRNA expressing vectors were fabricated using a Block IT miRNAi expression kit (Invitrogen, USA) following the supplied instructions. The kit had a control miR-expressing vector, i.e. the control miR. For crafting anti-miR-29a, oligonucleotides having at least 3–6 sequences complementary to miR-29a were infixed into a pmiR-report vector at the miR-29a decay site. The siRNA vectors were fabricated from the pSINsi-U6 DNA. The oligonucleotide was introduced in the mU6 DNA at the 5′-end of BamHI site and 3′-end of the Clal site. The oligonucleotides for the control siRNA and specific genes are described in Table I. The oligonucleotide constructs were infixed into the pLenti6/V5-D-TOPO vector, and the TR-β1 gene was cloned in the vector using the following primers: forward 5′-AGACAGAAAATGGCCTTCCAGCT-3′, reverse 5′-TAGTCCTCAAAGACTTCCAAGA-3′. Amplifying and cloning the 3′UTR region of the rat TR-β1 in reported luciferase was done by primers (Table II) following the supplied instructions.
DNA transduction and synthesis of lentivirus
The lentivirus stock was produced in cells suitable for lentiviral production; we used 293T cells for transfection. After 48 h of transfection the medium containing virus was collected and filtered using a 0.45 µm membrane filter. The lentiviral transfection was done by replacing the medium with medium containing virus supplemented with polybrene (5 µg/ml); the resultant was submitted to cooling and centrifuged at 10 000 rpm for 15 min at 15°C.
Extraction of RNA and qRT-PCR analysis
For qRT-PCR analysis, total RNA was isolated from neonatal rat ventricular myocytes and heart tissues of ICR mice or C57 black/6 Jacksonian mice; the isolation was achieved using TRIzol reagent. The cDNA from total RNA (5 µg) was synthesized using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher USA) following the supplied procedure. For qRT-PCR studies, genes were submitted to amplification (30 cycles) using Promega PCR master mix (Promega, USA). The expression was normalized using GAPDH as a loading control. The primers used for the analysis are described in Table III.
Table III
Primers used for qRT-PCR analysis
RNA blotting analysis
RNA blotting studies were done as reported earlier [21]. Briefly, from the total RNA a small fraction of RNA was isolated using a miR isolation kit (Thermo Fisher USA). About 5 µg of the RNA fraction was subjected to separation on a polyacrylamide gel (10%) by electrophoresis, then the resultant was transferred to a hybridization membrane using an electroblotter (Bio-Rad USA) at 4°C for 20 min. The RNAs were cross linked using 0.1 M each of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide and 1-methylimidazole for 2 h at 55°C at pH 8.4.
Luciferase reporter assay
For the luciferase assay, the oligonucleotide constructs were transfected into the 293FT cells using transfecting reagent (Thermo Fisher USA). The neonatal rat ventricular myocytes were transfected using Lipofectamine 2000 reagent. The transfection was done using the firefly luciferase reporter gene (0.1 µg), control plasmid Renilla reniformis (0.01 µg), and miR-RNAi expression vector (0.1 µg) for coding the specific miR or control. After 24 h of transfection, the luciferase activity was recorded using a dual reporter luciferase assay system. The results were normalized against luciferase activity of firefly.
Western blot analysis
Western blot analysis was done for studying the expression of proteins. For the study both incubated cells and heart tissues of C57 black/6 Jacksonian mice were homogenized in Tris buffer saline (pH 7.4). The buffer before use was supplemented with a protease inhibitor, 10 µM sodium orthovanadate and 0.5 mM sodium fluoride. The quantification of proteins was done by a protein estimation kit (Thermo Fisher USA). The proteins (2–10 µg) were fractionated on NuPAGE Novex 10% Bis-Tris protein gels (Thermo Fisher USA), then the gels were transferred to nitrocellulose membranes. The membranes were blocked with non-fat milk (5%) for 1 h followed by incubation with Iry antibody for 12 h at 4°C. The membranes were rinsed using 0.05% Tris-phosphate buffer saline, and the formed complexes were subjected to analysis. The Iry antibodies used for the experiments were anti-Dicer (dilution, 1 : 1000) Santa Cruz Biotech, anti-GAPDH (dilution, 1 : 1000) Santa Cruz Biotech, anti-αMHC (dilution, 1 : 20000) Abcam USA, β-MHC (dilution, 1 : 20000) Sigma Aldrich USA, anti-TR-β1 (dilution, 1 : 500) Santa Cruz Biotech USA and anti-RXRα (dilution, 1 : 500). The secondary antibodies used for the study were anti-mouse, anti-goat and anti-rabbit IgG at dilution 1 : 2000.
Results
Dicer siRNA suppresses miRNAs and α-MHC in neonatal rat ventricular myocytes
To study the effect of miRs on levels of MHC, we opted to suppress the function of endogenous miRs in neonatal rat ventricular myocytes. As we have elaborated about the importance of Dicer for synthesis of miRNA, it was speculated that the suppression of Dicer could lead to loss of function of miRNAs. The neonatal rat ventricular myocytes were subjected to transduction with siRNA with the aid of a lentiviral vector; which resulted in suppression of protein and Dicer mRNA under conditions in presence of serum (Figures 1 A, B).
Figure 1
Dicer siRNA suppresses the expression of endogenous miRs and α-MHC. The evaluation was done 72 h (A to C and E) or 96 h (D) after transfection with Dicer siRNA or control and miRs or miR-control in the NRVMs using lentiviral vector. The NRVMs used in the study were seeded under serum rich conditions. A, B – mRNA and protein levels of Dicer. C, D – qRT-PCR (C) and Western blot (D) analysis of mRNA and protein levels of α- and β-MHC. E – Results of qRT-PCR analysis for mRNA levels of α-MHC in NRVMs transduced with miRNAs compared to control miR
Data are presented as mean ± SE for n = 3. *P < 0.05, **p < 0.01 compared to respective controls.
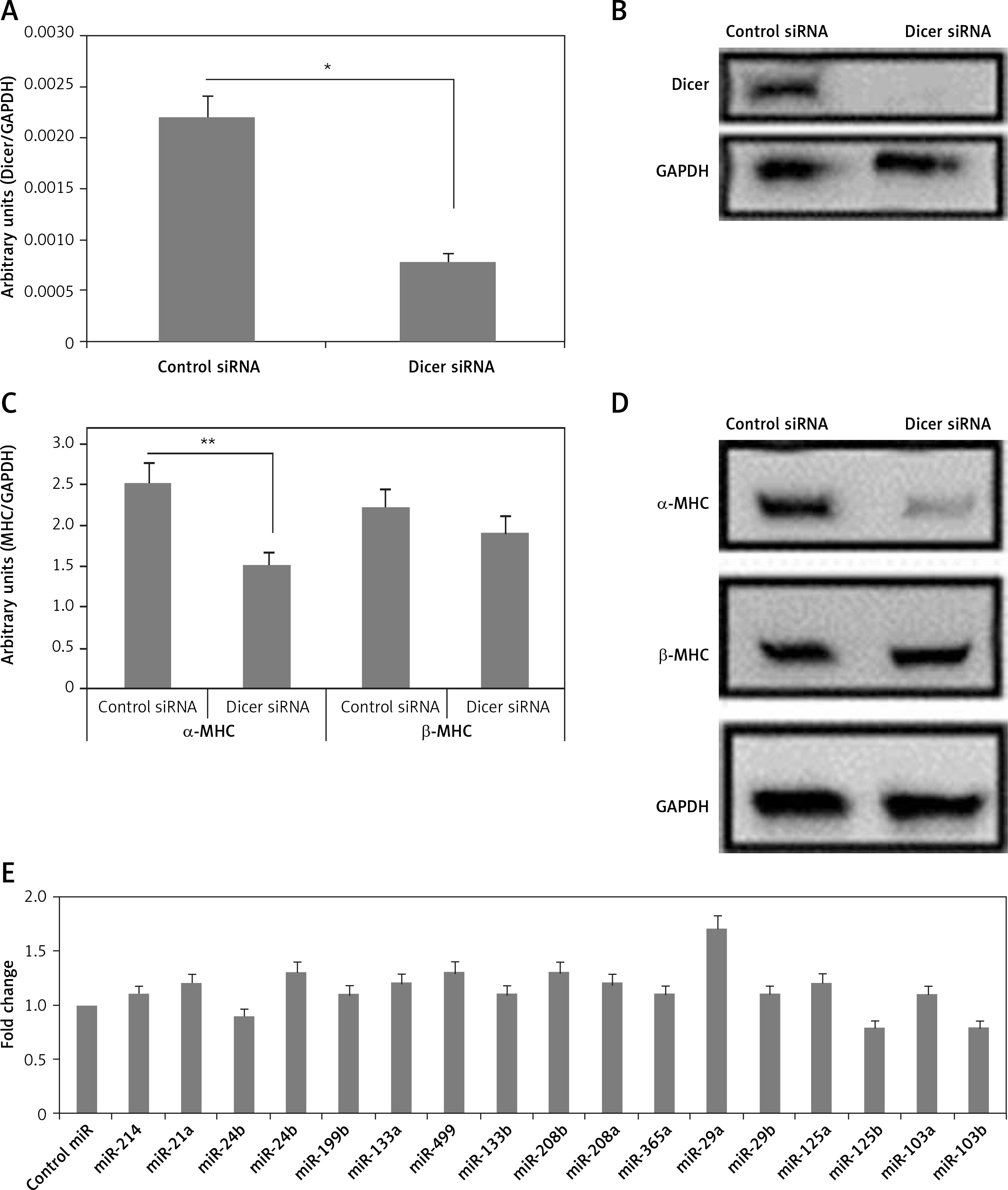
The expression levels of both α- and β-MHC were evaluated in neonatal rat ventricular myocytes transduced with Dicer siRNA. We observed that the transduction of Dicer siRNA downregulated the mRNA as well as protein levels of α-MHC (p < 0.01) but not of β-MHC compared to respective control siRNA (Figures 1 C, D). These findings suggested that the loss of function of miRs could lead to suppression of α-MHC in neonatal rat ventricular myocytes cultured with serum. We also found some specific miRs which can regulate the expression of α-MHC.
Next, to find the miRs that can increase the expression of α-MHC, 17 selected miRs were transduced individually in the neonatal rat ventricular myocytes, after which α-MHC mRNA levels were evaluated under serum containing conditions. Among the 17 selected miRs for the study, 5 were indentified earlier to be muscle specific (miR-208a, miR-208b, miR-133a, miR-133b and miR-499) [3, 12, 14, 22]. Additionally, 12 other miRs were included as they were reported to be over-expressed in some in vivo and in vitro cardiac models, malfunctioned cardiac tissues, cardiac failure and hypertrophy, and some miR microarray analysis also suggested them [18]. The results of qRT-PCR indicated multiple miRs increasing the levels of α-MHC but not significantly; however, we found that miR-29a increased the levels of significantly (Figure 1 E). The findings hence confirmed potential involvement of miR-29a in regulation of α-MHC under the present experimental conditions.
miR-29a up-regulates expression of α-MHC gene in neonatal rat ventricular myocytes
To find out whether miR-29a was over-expressed under altered cardiac conditions accompanied by the upregulated α-MHC gene, levels of miR-29a were evaluated in Dahl salt-sensitive rat hearts, which are reported to show high salt induced hypertensive hypertrophy after 11 weeks followed by cardiac failure at 17 weeks. It was observed that in the cardiac tissues of Dahl salt-sensitive rats given a high salt diet for eleven or seventeen weeks, the gene expression levels of α-MHC were upregulated along with upregulated mRNA levels of BNP and ANF (potential markers of cardiac pathology) (Figures 2 A, B). Additionally, miR-29a was over-expressed under low salt (LS) and high salt (HS) diet in cardiac tissues of Dahl salt-sensitive rat (Figure 2 C), demonstrating a link between miR-29a and α-MHC gene expression.
Figure 2
miR-29a increases expression of the α-MHC gene. A – Shows qRT-PCR analysis for levels of ANF, BNP and α-MHC, C – shows levels of miR-29a compared to GAPDH as loading control in heart tissues of salt-sensitive rats observed after 11 and 17 weeks. B – Western blot analysis for levels of α-MHC expression in low salt and high salt diet conditions. GAPDH was used as loading control. D – mRNA expression of α-MHC and DDR2 in fibroblasts and NRVMs; the values are mean and relative to untreated NRVMs. E – qRT-PCR analysis showing miR-29a expression levels in NRVMs and fibroblasts. F–H – After 72 h of transduction with miR-29a or control miR. F, G – miR levels of both α- and β-MHC. H – mRNA levels of ANF and BNP
The results are means ± SE (*p < 0.05, **p < 0.01, ***p < 0.001).
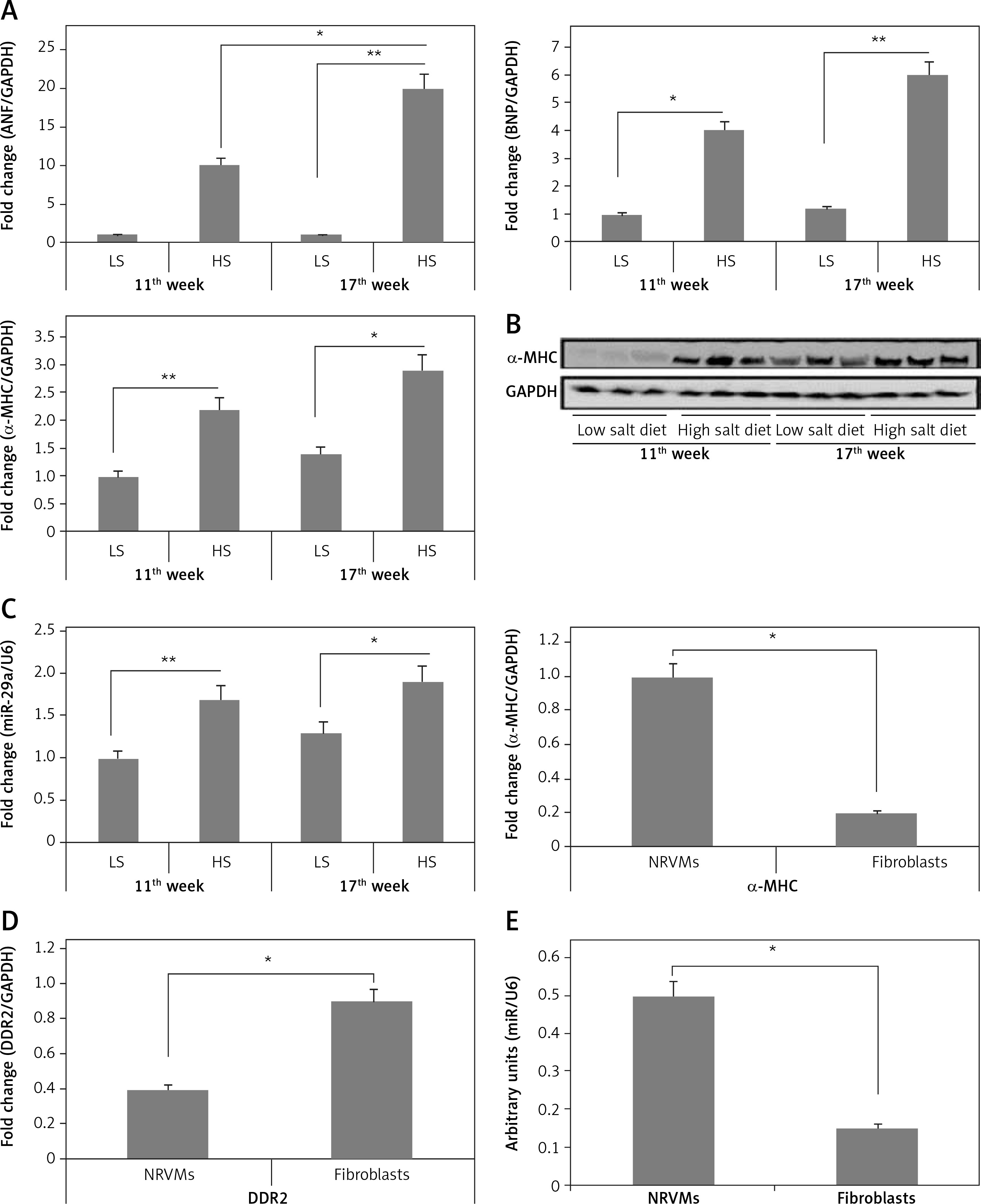
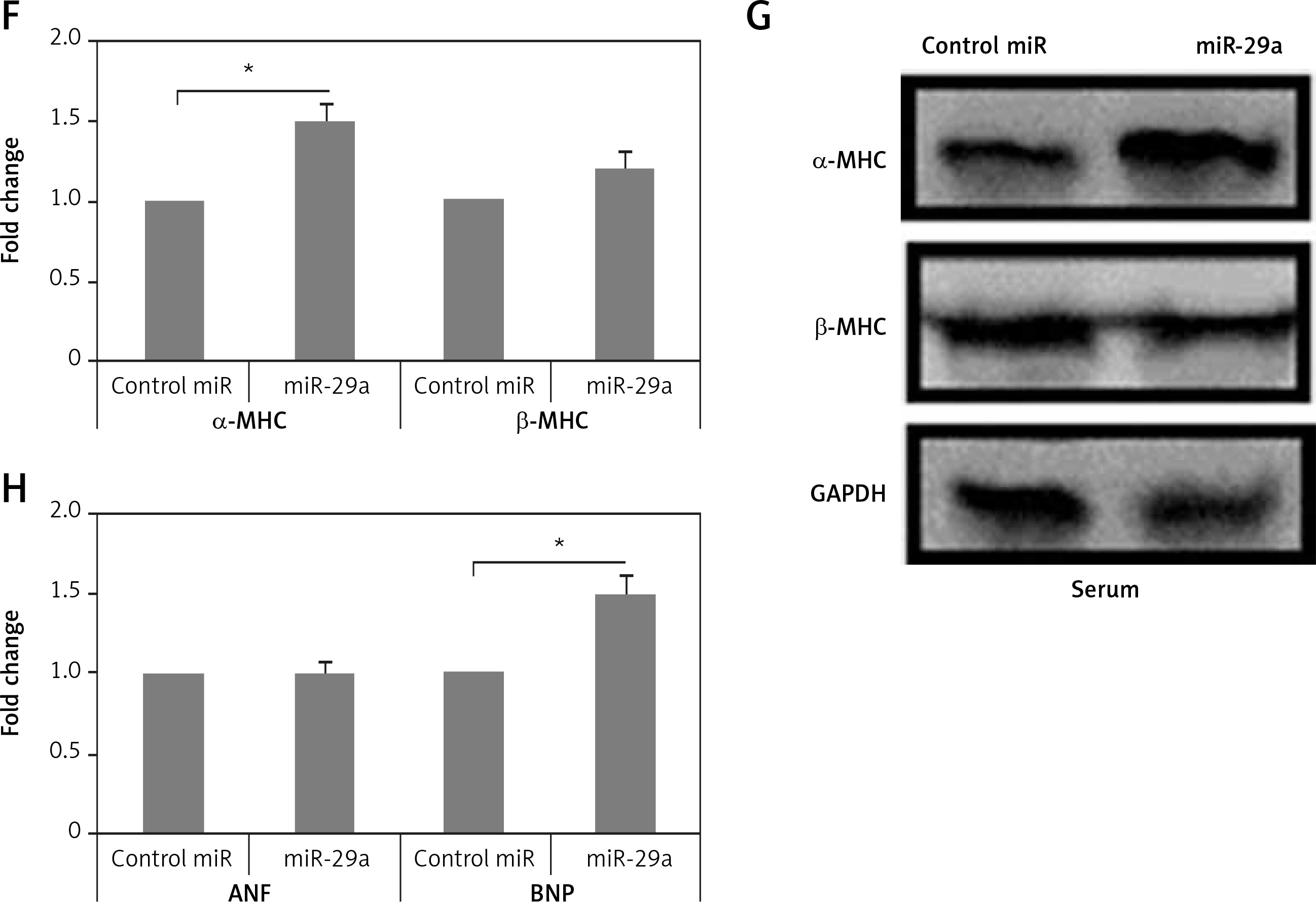
To evaluate whether miR-29a was expressed in cardiac myocytes endogenously, we assessed miR-29a levels in cardiac fibroblasts and neonatal rat ventricular myocytes. The optimum isolation of both types, i.e. fibroblasts and neonatal rat ventricular myocytes, was confirmed by evaluating the mRNA levels of α-MHC and discoidin domain receptor 2 (DDR2) (Figure 2 D). We found that miR-29a was expressed significantly more highly (p < 0.05) in neonatal rat ventricular myocytes compared to the fibroblasts (Figure 2 E). Also over-expression in myocytes affirmed that miR-29a could lead to upregulation of α-MHC significantly (p < 0.05) and not the β-MHC gene at protein as well as at mRNA levels under serum enriched conditions (Figures 2 F, G). miR-29a could also up-regulate the mRNA levels of BNP but not of ANF (Figure 2 H). The results hence suggested that the miR-29a mediated upregulation of the α-MHC gene may be a direct effect of miR-29a rather than due to an IIry change to heart pathology induced by miR-29a.
Non-functioning miR-29a decreases α-MHC protein levels in neonatal rat ventricular myocytes
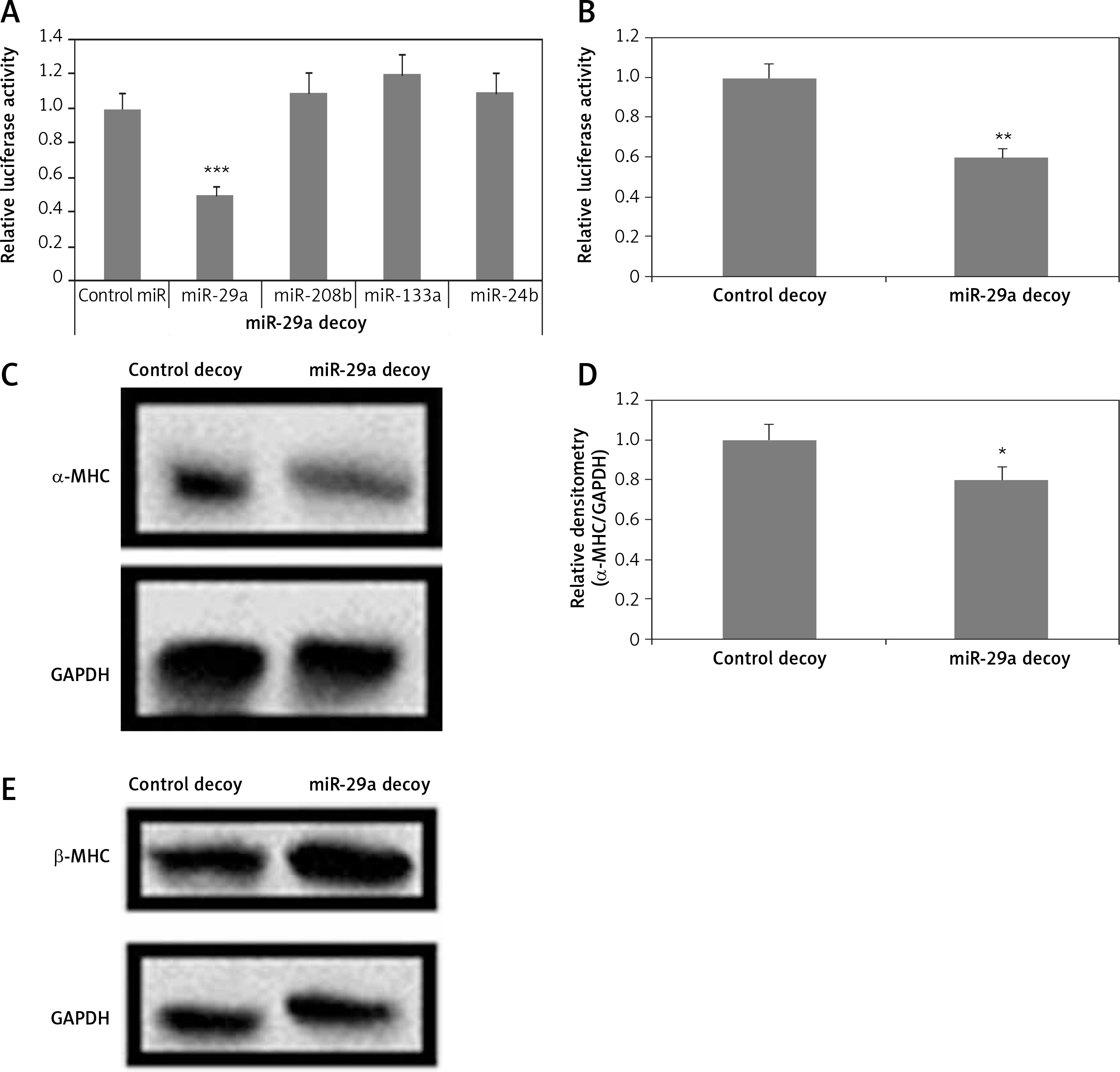
To evaluate the direct consequences of miR-29a on regulation of the α-MHC gene, the protein levels of α-MHC were evaluated in neonatal rat ventricular myocytes in which the functioning of miR-29a was suppressed. The neonatal rat ventricular myocytes were transfected with a lentiviral vector having a 3′UTR region with 3 to 6 tandem miR-29a decoy sequences; these were complementary with the sequence in miR-29a linked to a luciferase reporter gene as reported earlier [23–25]. The decoy sequences parted the endogenous miR-29a. When miR-29a was transfected in the 293T cells using a miR-29a decoy, it resulted in decrease of luciferase activity significantly compared to control miR (p < 0.001) (Figure 3 A). It was also demonstrated that miR-208b, miR-133a and miR-24b did not affect the luciferase activity (Figure 3 A). Then, the neonatal rat ventricular myocytes were transfected with miR-29a decoys; this transfection caused decrease in luciferase activity significantly compared to the control decoy (p < 0.01) (Figure 3 B). These findings suggested that miR-29a decoys could bind specifically to the endogenous miR-29a. We also found that upon transducing miR-29a decoys in the neonatal rat ventricular myocytes using lentiviral vectors, α-MHC protein levels were down-regulated, but the levels of β-MHC remained unaltered under serum enriched conditions (Figures 3 C–E). These results confirmed that miR-29a is involved in regulation of the α-MHC gene in neonatal rat ventricular myocytes. MiR-208a and miR-499 are reported to be inducers of MHC [3, 14]. To evaluate the involvement of inducing miRs in regulation of α-MHC a study similar to miR-29a was done using the miR-208a decoy. The miR-208a decoy binds to endogenous miR-499 and miR-208a (Figures 3 A, B). A decoy works by binding a specific miR which is complementary to the sequences in the decoy; miR-499 and -208a have been found to have similar sequences and could bind the miR-208a decoy [25].
Figure 3
miR-29a decoy suppresses protein levels of α-MHC. A – 293T cells received transfection of luciferase miR-29a decoy construct along with miR plasmid or control-miR. B – miR-29a and control decoy were transfected in the NRVMs; the results are relative to control. C–E – Assays were done 72 h post transfection. The NRVMs were transduced with miR-29a decoy and protein expression of α- and β-MHC was evaluated
The results are means ± SE (*p < 0.05, **p < 0.01, ***p < 0.001).
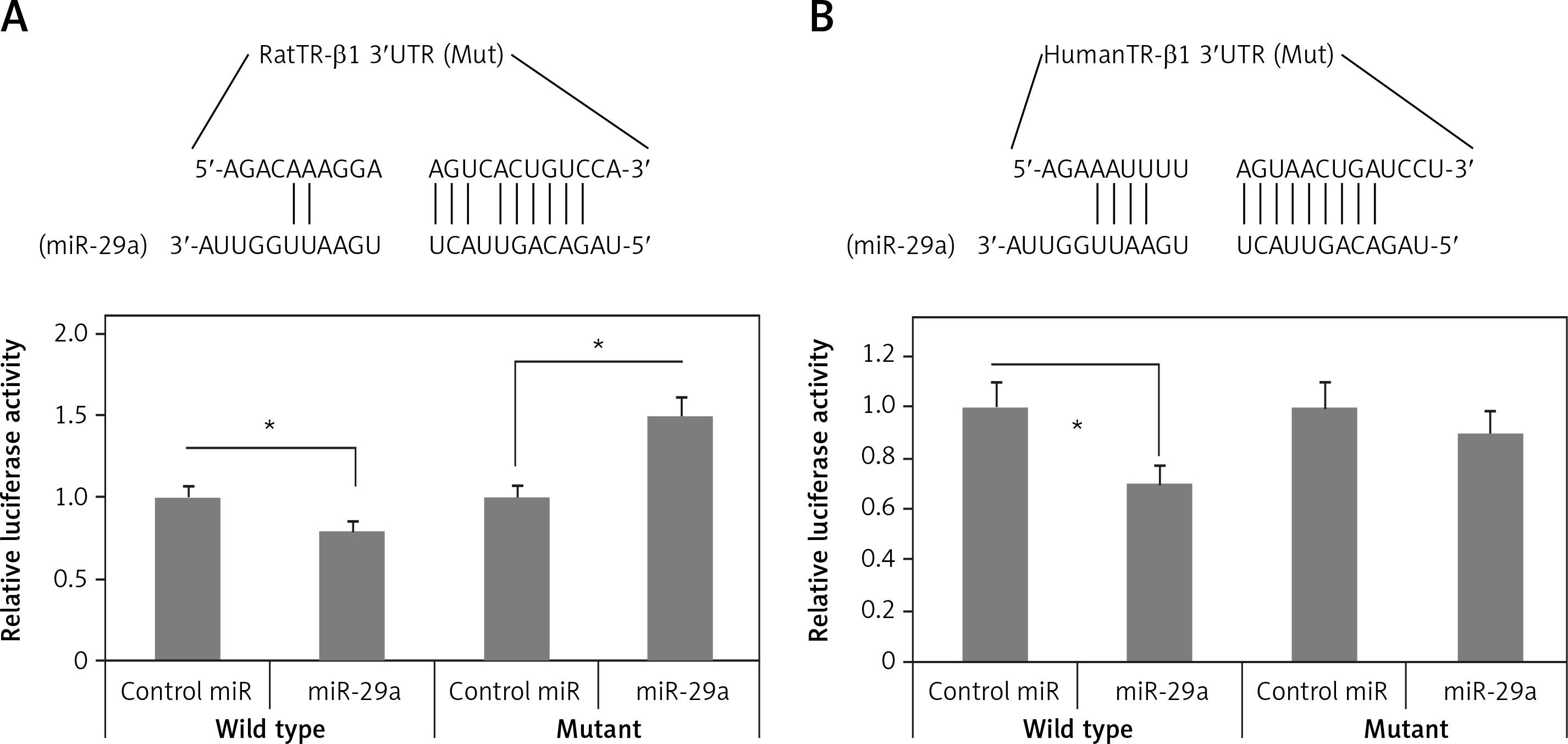
Bioinformatics studies for prediction of target of miR-29a
To identify the targets of miR-29a which can regulate the expression of α-MHC, bioinformatics analysis was done by TargetScan for identifying potential targets of miR-29a. We speculated that upregulation of miR-29a could suppress the target gene which can regulate the expression of α-MHC negatively; this might lead to upregulation of α-MHC. It was expected that the transcription factors responsible for negative regulation of α-MHC or its associated co-regulators would be involved in upregulation of α-MHC. Literature has suggested multiple factors which can bind to the rat α-MHC promoter [26], TargetScan predicted TR-β1 to be one of the targets of miR-29a (Figure 4 A). TR-β1 negatively regulates the transcription of α-MHC by binding to thyroid hormone response element (TRE) [27]. Further, the 3′UTR of TR-β1 was transfected into the luciferase construct which was transduced into 293T cells and the cytomegalovirus miR-29a decreased the luciferase activity of the construct (Figure 4 A). The results also demonstrated that the luciferase activity decreased significantly for TR-β1 transfected 3′UTR reporter (p < 0.05) (Figure 4 B). It was however noted that the luciferase activity remained unaffected in the mutant reporters by miR-29a (Figures 4 A, B). These results demonstrated that the binding sites of miR-29a in the 3′UTR region of TR-β1 could lead to translational suppression due to miR-29a.
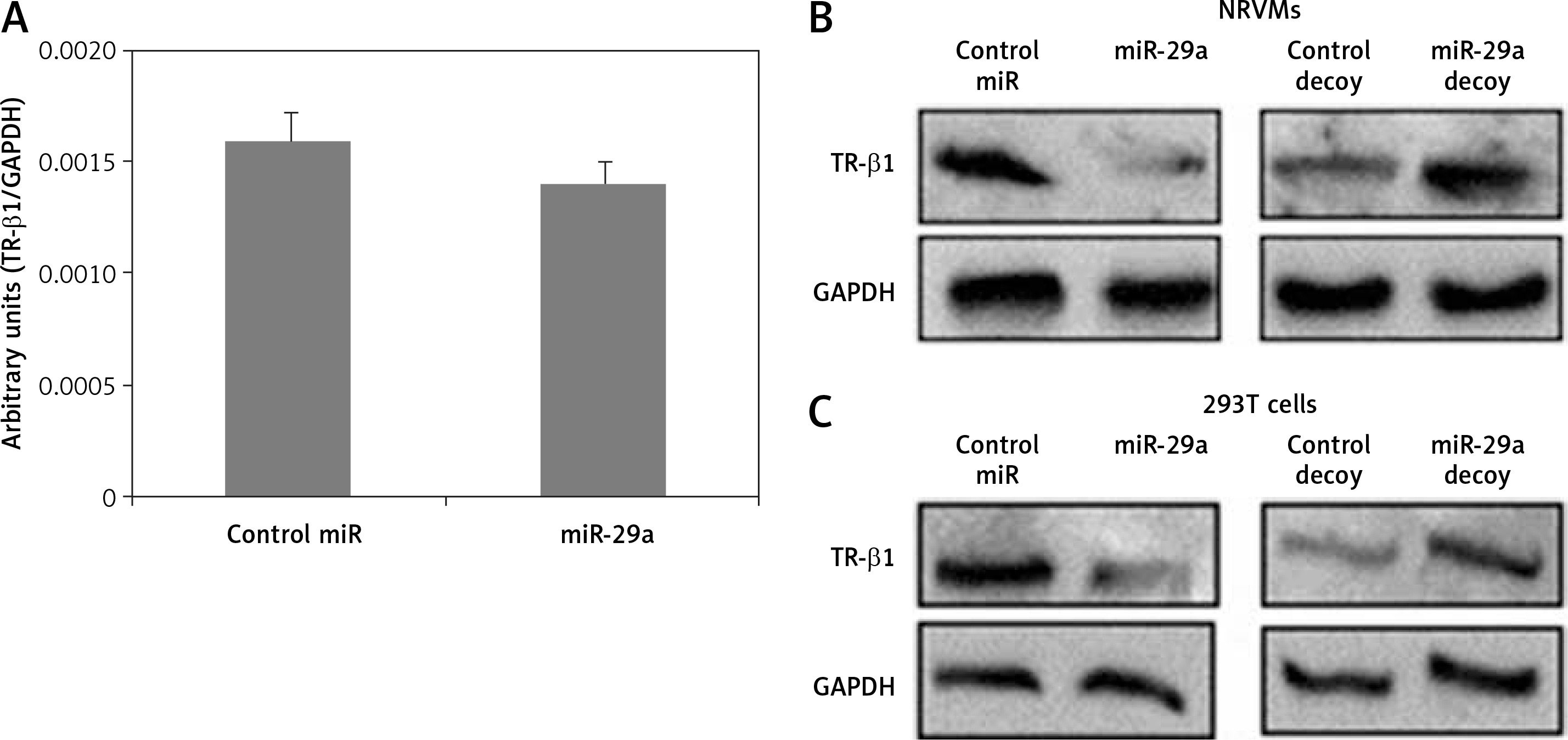
Thyroid hormone receptor-β1 is a potential target of miR-29a
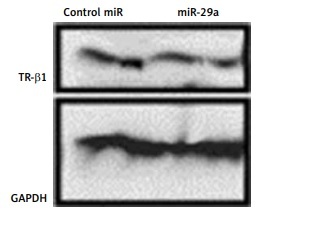
To confirm whether miR-29a specifically targets TR-β1, the expression of TR-β1 was studied in neonatal rat ventricular myocytes or 293T cells transfected with miR-29a or its decoy. We found that miR-29a down-regulated the protein levels of TR-β1 without altering the mRNA levels in the neonatal rat ventricular myocytes. We also found that the miR-29a decoy enhanced the protein levels of TR-β1 (Figures 5 A, B). Similar results were observed in the case of 293T cells (Figure 5 C). These outcomes suggested that TR-β1 is a favorable target of miR-29a in human as well as in rat cells.
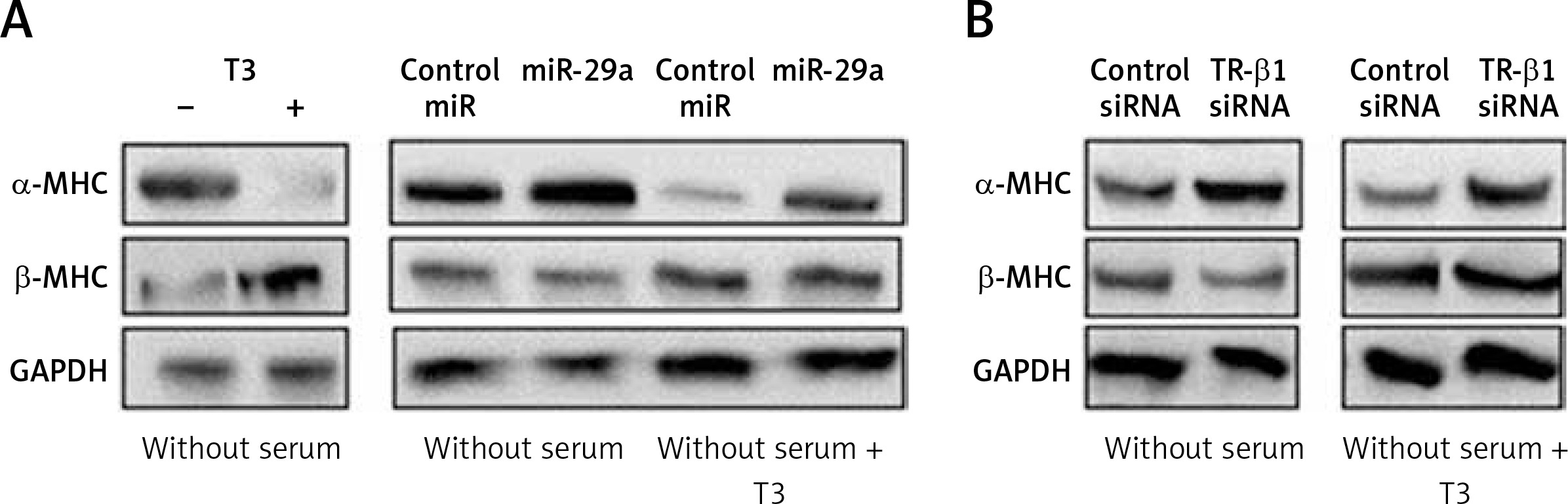
miR-29a regulates levels of α-MHC protein via TR-β1
To evaluate whether miR-29a is involved in regulation of α-MHC expression through TR-β1, we subjected the neonatal rat ventricular myocytes to miR-29a or its decoy followed by treatment with tri-iodothyronine (T3) and the protein levels of α-MHC were assessed. We found that the treatment of T3 (10 nM) in neonatal rat ventricular myocytes upregulated the expression of α-MHC significantly and down-regulated the levels of β-MHC (Figure 6 A). These findings were in agreement with a study reported earlier in which T3 induced the expression of α-MHC in cardiac myocytes [29]. Up-regulated miR-29a attenuated the effect of T3, which caused an increase in protein levels of α-MHC but did not alter the expression of β-MHC (Figure 6 A). The results after TR-β1 siRNA treatment were also similar (Figure 6 B), hence suggesting regulation of the α-MHC gene by miR-29a through TR-β1.
Up-regulation of miR-29a increases the α-MHC gene in mice ES cells
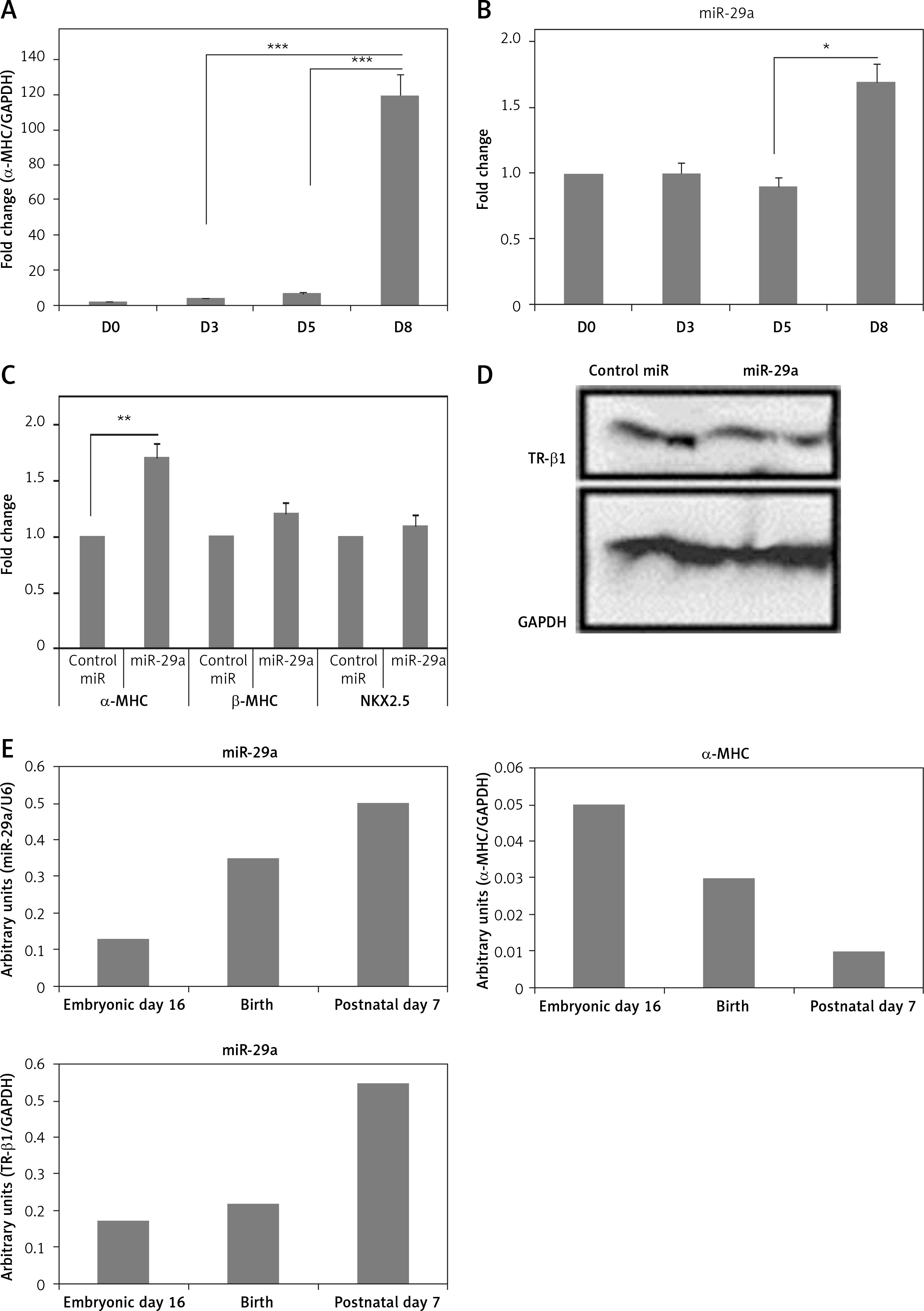
To study the physiological effect of miR-29a on TR-β1-mediated regulation of α-MHC in mice, we evaluated the expression of miR-29a and MHC in cardiac tissues of mice ES cells at the stage of differentiation into cardiomyocytes. The ES cells were incubated on gelatin-coated plates and allowed to differentiate for 8 days. The mRNA levels of α-MHC in mice ES cells increased significantly between day 5 and 8 (Figure 7 A) [28], whereas the levels of miR-29a remained unchanged until the 5th day, but increased up to 1.5 times in between the 5th and the 8th day (Figure 7 B). Additionally, over-expression of miR-29a in ES cells of mice increased the mRNA levels of α-MHC, but failed to alter expression β-MHC or NKX2.5, which is an indicator of cell differentiation of cardiac cells, and decreased the protein levels of TR-β1 (Figures 7 C, D). These outcomes suggested that the over-expression of miR-29a could up-regulate levels of α-MHC via TR-β1 without affecting the cell differentiation of mice ES cells. To investigate the physiological role of miR-29a in development of cardiac tissues, the levels of miR-29a, α-MHC and TR-β1 were studied. The outcomes demonstrated that, during the process of cardiac development, the mRNA levels of miR-29a and TR-β1 increased progressively, whereas the levels of α-MHC decreased (Figure 7 E).
Figure 7
Up-regulation of miR-29a increases the gene expression of α-MHC in mouse ES cells. qRTPCR analysis of α-MHC mRNA (A) or miR-29a levels during the differentiation of ES cells in mouse (B). Both the levels of α-MHC mRNA and miR-29a were represented relative to levels at day 0. C – qRTPCR analysis for mRNA levels of α- and β-MHC and NKX2.5 were presented. D – protein levels of TR-β1 by western blot analysis in ES cells from mouse at the eighth day after transduction with miR-29a or control miR. E – qRT-PCR analysis of mRNA levels of miR-29a or α-MHC and TR-β1 in heart tissues at the sixteenth day in the embryonic mouse, at the birth and at postnatal day 7
The results are means ± SE (*p < 0.05, **p < 0.01, ***p < 0.001).
miR-29a is over-expressed in cardiac tissues of mice exposed to TAC
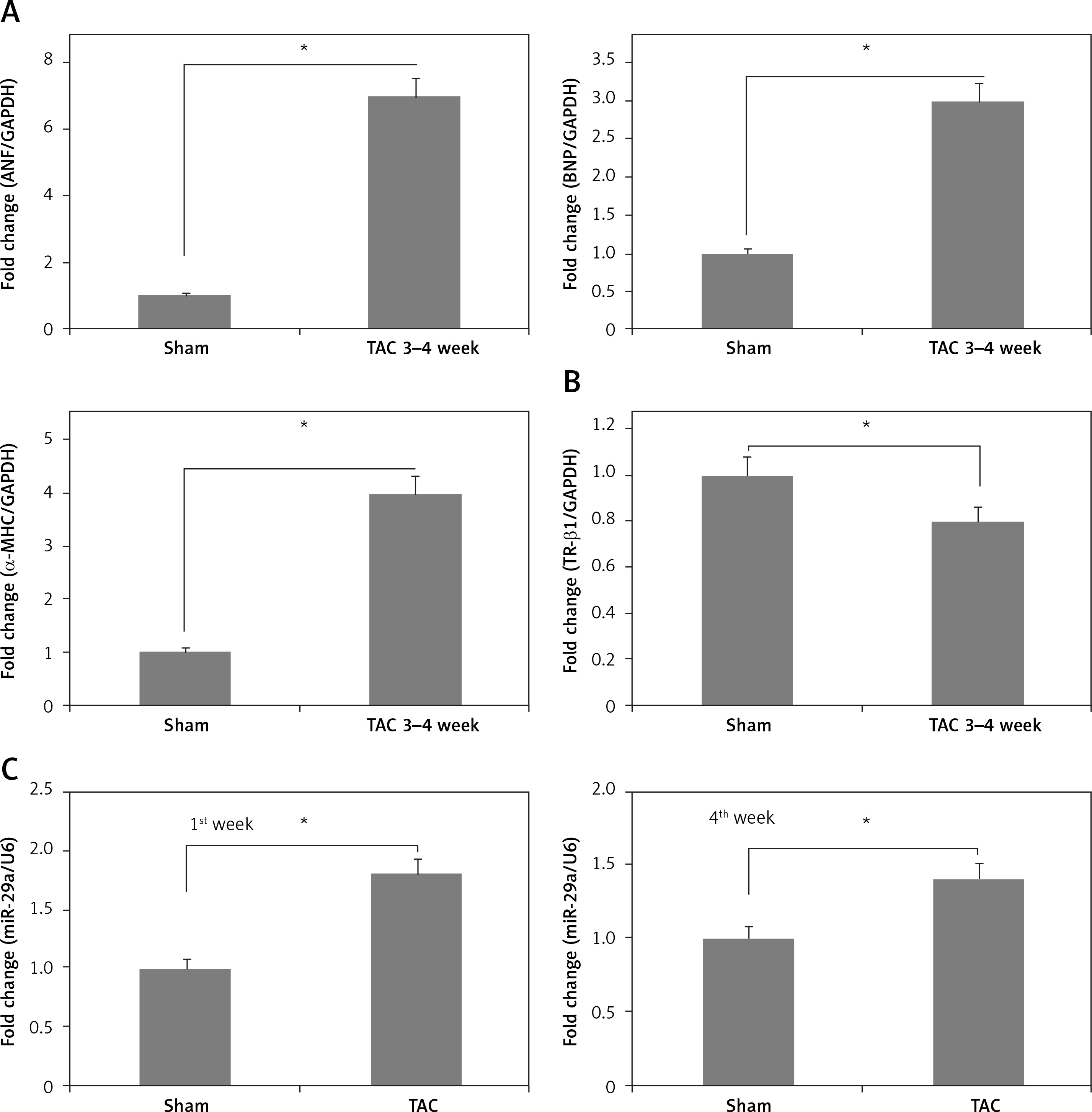
To confirm the role of miR-29a in vivo, the expression levels of miR-29a, α-MHC and TR-β1 were evaluated in cardiac tissues of mice treated with TAC, which causes cardiac hypertrophy in mice. The results demonstrated that expression of α-MHC increased and that of TR-β1 was decreased during the time interval between the 3rd and 4th week of TAC exposure (Figures 8 A, B). Additionally, expression of miR-29a was up-regulated during the 1st and 4th week of TAC treatment (Figure 8 C). These outcomes indicated that expression of miR-29a could be linked to development of cardiac hypertrophy via regulation of α-MHC through TR-β1.
Figure 8
miR-29a is over-expressed in the heart tissue of mice treated with TAC. qRT-PCR analysis showing mRNA expression of ANF, BNP and α-MHC (A), TR-β1 mRNA (B) in heart tissues of mice at 3–4 weeks after TAC. C – Fold changes in expression of miR-29a in heart tissues of sham and TAC mice at 1 week and 4 weeks after TAC
The results are means ± SE (*p < 0.05, **p < 0.01, ***p < 0.001).
Discussion
A number of studies have investigated the role of miRs in the cardiovascular system. However, it is difficult to identify a target specific miR from multiple miRs as these micro-molecules can alter several target genes simultaneously. In in vivo analysis, a number of factors such as hemodynamics, cytokines and hormones can affect gene regulation in cardiac cells, which makes the process of evaluating the effect of a specific miR difficult. In the present study, we attempted to evaluate the direct effect of miRs on regulation of cardiac genes using the Iry cultures of neonatal rat ventricular myocytes. We selected neonatal rat ventricular myocytes as these cells have a minimal effect of environmental factors or any possible interactions from the other cellular components.
In the present study, we attempted to suppress the miRNA function neonatal rat ventricular myocytes via siRNA against an enzyme named Dicer which is necessary for biosynthesis of miR. We also studied the effects of target miRNA on expression levels of a cardiac specific gene, i.e. MHC.
Earlier in a study, targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure by targeting α-MHC [29]. The outcomes of the present study suggested that Dicer siRNA leads to down-regulation of α-MHC. These variations in outcomes may be due to differences in expression of α-MHC in the perinatal and fully grown mice or may be from the hormonal and the hemodynamic effects in vivo [30].
The current findings demonstrate a novel role of miR-29a as an activator of the α-MHC gene. miR-208a and miR-499 were identified as established activators of MHC genes [24, 25]; here the transduction showed no significant alterations in expression of α-MHC; the reduction of miR-29a levels was significant compared to that shown by miR-208a and miR-499 in neonatal rat ventricular myocytes transfected with Dicer siRNA. Additionally, the miR-29a decoy decreased the protein levels of α-MHC, whereas the decoy of miR-208a failed to do the same. Hence the outcomes confirmed that the effects of miR-29a were significant compared to those of miR-208a and miR-499, which are muscle-specific miRNAs and are known to play important roles in regulation of MHC levels [25].
In the present study we found that miR-29a could be a modulator of the thyroid hormone receptor pathway in the α-MHC gene. Also the expression of MHC gene is potentially modulated by thyroid hormone and receptors. The results suggested that TR-β1 regulates the expression of α-MHC in neonatal rat ventricular myocytes, which was in agreement with an earlier study [31]. The present results also suggested that levels of β-MHC remained unaffected by TR-β1 in neonatal rat ventricular myocytes. These results suggested that miR-29a can regulate the thyroid hormone pathway by regulating the α-MHC gene via TR-β1 in neonatal rat ventricular myocytes. We also evidenced that in absence of triiodothyronine, the upregulation of TR-β1 or the miR-29a decoy caused no significant decrease in mRNA or protein levels of α-MHC. Previously it has been evidenced that levels of α-MHC can be suppressed by over-expression of TR-β1 [31].
In the present study, we showed that in absence of tri-iodothyronine, TR-β1 up-regulation suppressed the luciferase activity of the α-MHC construct, having the α-MHC promoter sequence, but did not suppress the α-MHC protein and mRNA levels. It was speculated that there may be some other factors which may up-regulate the expression of α-MHC in the absence of triiodothyronine outside the promoter region of α-MHC involved in the study. We observed that in serum or triiodothyronine deprived conditions, TR-β1 showed a weak effect on α-MHC, whereas in serum enriched conditions TR-β1 changed the α-MHC levels significantly, which suggested that some factors present in serum could regulate the expression of the α-MHC gene via TR-β1. The medium enriched with serum was found to have tri-iodothyronine at levels ranging from 1 to 0.1 nM. As per the reports published earlier, at levels of 1 nM tri-iodothyronine could alter the mRNA levels of α-MHC [31]. These results indicate that the presence of tri-iodothyronine in serum could regulate the expression of α-MHC by miR-29a; additionally there may be a possibility of some other factors being involved in regulation of the α-MHC gene.
In the present study, the role of miR-29a was also elucidated in an animal model. It was evidenced that at the stage of ES cell differentiation, the expression of miR-29a increased along with α-MHC mRNA and was associated with down-regulation of protein levels of TR-β1. Additionally, miR-29a was over-expressed in cardiac myocytes [32]. This finding means that miR-29a could be associated with regulation of α-MHC during differentiation of ES cells, which may play an important role in development of the heart, also indicating the involvement of factors other than miR-29a in regulation of TR-β1 during the process of ES differentiation to cardiac cells in development of cardiac tissues.
In this study, using the TAC-induced C57 black/6 Jacksonian mice we found that both miR-29a and α-MHC were over-expressed along with down-regulation of TR-β1. The up-regulation of miR-29a in neonatal rats’ ventricular myocytes seemed to elevate the expression of α-MHC directly rather than inducing cardiac pathological conditions. These outcomes suggested that over-expression of miR-29a was stress-induced, through cardiac hypertrophy and increased levels of α-MHC via TR-β1. However, it was still unclear whether the deregulation of TR-β1 affected the heart pathophysiology, as the TR-β1 deficient mice have been reported to possess normal cardiac function [33]. However, the TR-β1 knockout mice do not bind with tri-iodothyronine associated with decreased contractile function [34]. Hence, the tri-iodothyronine pathway through the TR-β1 may be important under the stress conditions and the over-expression of miR-29a in mice might affect the function of the heart. Further evaluation is needed to determine the precise functioning of TR-β1 in cardiac pathophysiology.
In conclusion, miR-29a can modulate the expression of α-MHC by targeting TR-β1 in neonatal rats’ ventricular myocytes. Also the over-expression of miR-29a in cardiac hypertrophy appears to increase expression of α-MHC through TR-β1.