Introduction
Natural products have been one of the main sources of inspiration for new anti-cancer drugs [1]. Withaferin-A (WA), extracted from the Indian medicinal herb Withania somnifera, has been proven to have anti-tumor and anti-inflammatory effects [2] in various cancer cells, including colorectal cancer (CRC), ovarian cancer, breast cancer, leukemia, glioblastoma, neuroblastoma, multiple myeloma, and head and neck cancer [3–5]. WA involves multiple molecular mechanisms, including targeting the proteasome system and cytoskeleton, and regulating HSP90 activity of heat shock protein 90 (HSP90), inhibiting nuclear factor κB, and reactive oxygen species-mediated cytotoxicity, etc. [6]. Jan et al. [7] reported that WA inhibits hypoxia-inducible factor-1α of non-small cell carcinoma, thereby inhibiting its metastasis. This finding shows the potential of WA as an anti-cancer drug. Currently, the impact of WA on CRC remains unclear. The present study aimed to explore the effect of WA on CRC. Our research showed that WA can inhibit the growth and metastasis of CRC in vitro and in vivo, with transcriptome sequencing showing that WA inhibited the EMT pathway, HSP90, and HIF-1α genes of CRC. Expression of hypoxia-inducible factor-1α (HIF-1α) was elevated in cells during hypoxic stress. Being a key transcription factor, HIF-1α is found in cancer glycolysis, angiogenesis, and cancer metastasis. It plays a role in promoting cancer metastasis as an important molecule in the EMT pathway. It is also a client protein of HSP90 [7].
Epithelial-mesenchymal transition pathway (EMT) is a crucial step in the invasion and metastasis of colorectal cancer cells, and provides the environment for cancer cells to survive hematogenously, ultimately affecting drug resistance [8]. In addition, EMT allows cells to break away from the initial lesion and form new metastases elsewhere, which is known to be an important process of hematogenous dissemination [9, 10]. Hence, suppressing EMT might be a good strategy to tackle cancer treatment.
Our study demonstrated the anti-tumor effect of WA in CRC in both vivo and vitro conditions. We also revealed the underlying mechanism. These findings may provide a valuable theoretical basis for future CRC treatment studies.
Material and methods
Extraction of WA
WA was purchased from HerbPurify Co., Ltd (Chengdu, China). It was identified and confirmed by mass spectrometry (Supplementary Figure S1). A stock solution of WA (5 mM) was prepared in dimethyl sulfoxide (DMSO) and stored in aliquots at –20°C until use.
Cell culture
HCT116 and SW480 cells used in this study were ordered from Kegen Biotechnology Co., Ltd (Nanjing, China). These cell lines were cultured in RPMI-1640 (Gibco, Grand Island, USA) supplemented with 10% fetal bovine serum (HY, USA) in 5% CO2. The medium was changed every 48 h, and cells were passaged at 80% confluence for less than 20 cycles.
HIF-1α-targeting shRNA (short hairpin RNA) and lentivirus packaging
Lentivirus packaging was completed as described in a previous study [11]. Lipofectamine 2000 (Life Technologies, California, USA) was used for shRNA transfection according to the protocol provided by the manufacturer. To establish stable HIF-1α knocked down cell lines, we cloned oligonucleotides encoding short hairpin RNA (shRNA) into lentiviral vector psi-LVRU6GP (Genecopeia), which were named sh-1 and sh-2, respectively. A scrambled shRNA named sh-Ctrl was used as a negative control. Sequences of sh-1, sh-2, and sh-Ctrl were as follows: CCGCTGGAGACACAATCATAT, CCAGTTATGATTGTGAAGTTA and GTGATGAAAGAATTACCGAAT. After being transfected with sh-RNAs (sh-1, sh-2, or sh-Ctrl), CRC cells were later cultured in puromycin-supplemented medium (2 µg/ml puromycin) for 4 weeks to establish stable cell lines. To construct CRC cells in which HSP90 and HIF-1α were overexpressed, we then transfected HSP90-overexpression-promoting plasmid (GVHSP90), HIF-1α-overexpression-promoting plasmid (GV HIF-1α) and negative control plasmid (GV-Vector) with GV492 as the carrier, which were designed and synthesized by GeneChem (Shanghai, China), into CRC cells with Lipofectamine 3000 (Invitrogen, Carlsbad, USA).
Cell viability and colony formation assay
Cells were seeded into a 96-well transparent bottom black board with 3,000 cells per well during the cell proliferation assay. WA was added to each well at different final concentrations (0, 0.05, 0.1, 0.2, 0.4, 0.8 µM) for 24 h to determine the concentration. Cell viability was examined by spectrophotometry at 450 nm wavelength after CCK8 reagent (CCK8, Dojindo, Japan) was added 2 h later. It was then placed in a humidified incubator containing 5% CO2 and 95% air. Subculture and all experiments were performed at 37°C.
To further determine the effect of WA on CRC cell tumourigenicity, colony formation assay was performed. HCT116 and SW480 cells were separately seeded in 6-well plates with 500 cells per well during the colony formation assay. WA was added to each well at specified concentrations (0, 0.05 and 0.1 µM) after we had determined the proper concentration after the colony formation, and then the cells were incubated for 12 days. The cells were fixed with 4% paraformaldehyde for 30 min, and later stained with crystal violet and counted under a microscope.
Cell migration and invasion assay
To evaluate the effects of WA on CRC cell metastasis, transwell assay was performed. In migration assay, the upper chamber (8-µm pore size) containing serum-free medium was seeded with 5 × 104 cells, while the lower chamber was permeated with 20% FBS-supplemented medium. HCT116 and SW480 cells were incubated for 38 h and 45 h [12], respectively, and then fixed with 4% paraformaldehyde. The remaining cells were swabbed off after the incubation. Cells that had crossed the membrane were then stained with crystal violet. After rinsing off the excess dye, the cells that had passed through the membrane were counted and averaged.
In the matrix invasion assay, the Matrigel (Corning) was diluted with PBS at a ratio of 1 : 8. It was solidified in a 37°C incubator for 15 min. The rest of this assay was identical to the migration assay.
Biochemistry and immunohistochemical staining
Mice in the subcutaneous xenograft model were sacrificed, and their serum, kidney, liver and heart were collected at the end of the experiment to evaluate the toxicity of WA. Serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured by a fully automatic biochemical analyzer (TC9080, TECOM). Kidney, liver and heart were stained with HE and then imaged under a microscope.
In the immunohistochemical staining assay, all the animal tissue samples were initially fixed in 10% neutral buffered formalin, and later processed by dehydration and paraffin embedding. The samples were then sectioned at a 3-µm thickness. According to the protocols provided by the manufacture, we performed immunohistochemical (IHC) staining utilizing these fixed and paraffin-embedded tissues with an Immunohistochemistry Kit (Sangon Biotech, Shanghai, China). Primary antibodies (HSP90 (1 : 5000, Proteintech, Cat No. 60318-1-Ig) and HIF-1 a (1 : 1000, Proteintech, Cat No. 20960-1-AP) were used for the IHC staining. Horseradish peroxidase-conjugated streptavidin was later introduced for 30 min. Positive cells were counted using different fields under a microscope after DAB staining. Staining intensity score of this study was defined according to the formula as described in the literature [13].
Quantitative real-time PCR
Predetermined concentrations of WA were used to incubate the cells for 12 h. Trizol reagent (Accurate Biology, Changsha, China) was used to extract total RNA according to the protocols provided by the manufacturer. qRT-PCR was performed according to the protocols provided by the manufacturer using Evo M-MLV reverse transcriptase premix and the SYBR Green Premix Pro Taq HS qPCR kit (Accurate Biology). The relative expression of E-cadherin, vimentin, Hsp90, HIF-α and GAPDH mRNA was determined after normalization. The primers used are listed below, and the fold change of each sample was determined by the 2–ΔΔCt relative quantification method. E-cadherin, forward primer: 5ʹ-CCACCAAAGTCACGCTGAAT-3ʹ, reverse primer: 5ʹ-GGAGTTGGGAAATGTGAGC-3ʹ. Vimentin, forward primer: 5ʹ-GAGAACTTTGCCGTTGAAGC-3ʹ, reverse primer: 5ʹ-CTCAATGTCAAGGGCCATCT-3ʹ. GAPDH, forward primer: 5ʹ-GACAGTCAGCCGCATCTTC-3ʹ, reverse primer: 5ʹ-CAACAATATCCACTTTACCAG-3ʹ. Hsp90, forward primer: 5ʹ-CGATGAATATGCCATGACT-3ʹ, reverse primer: 5ʹ-TCCATAGCAGATTCTCCAG-3ʹ. HIF-α, forward primer: 5ʹ-TGATGTGGGTGCTGGTGTC-3ʹ, reverse primer: 5ʹ-TTGTGTTGGGGCAGTACTG-3ʹ.
Western blotting
We extracted the total protein from cells on ice. Protein concentration was measured, then protein was separated using the SDS-PAGE method (Beyotime) at 95°C for 8 min and transferred onto PVDF membranes (Millipore, Billerica, USA). These PVDF membranes loaded with the transferred proteins were soaked in 5% BSA solution for at least 1 h, then immediately incubated at 4°C overnight with different primary antibodies. On the second day, the PVDF membranes were washed with TBST solution three times and later again with secondary antibodies at room temperature for a minimum of 1 h. After a 30-minute wash with TBST solution, the antibody-conjugated protein bands on the PVDF membranes were visualized under an imaging system (Bio-Rad ChemiDoc MP) with a BeyoECL Plus Kit (Beyotime Biotechnology). Information regarding antibodies is as follows: HSP90 (1 : 5000, Proteintech, Cat No. 60318-1-Ig), HIF-1α (1 : 1000, Proteintech, Cat No. 20960-1-AP), anti-GAPDH rabbit polyclonal antibody (1 : 5000, Proteintech, Cat No. 60004-1-Ig).
Immunoprecipitation
To evaluate the interaction effect of WA on HSP90 and HIF-1α, immunoprecipitation assay was performed. After cell lysis using RIPA Lysis Buffer (Beyotime, Shanghai, China) and centrifugation, the supernatant was collected. Protein concentration was measured for 2,000 µg of protein lysate using the BCA Protein Assay Kit (Beyotime). Then antibody was added to lysates with protein A beads (Thermo Fisher Scientific, CAT#20424) and left to incubate overnight. The beads were then Western blotted. Information regarding antibodies is as follows: HSP90 (2.0 µg for 2.0 mg of total protein lysate, Proteintech, Cat No. 60318-1-Ig), HIF-1α (2.0 µg for 1.0 mg of total protein lysate, Proteintech, Cat No. 20960-1-AP), anti-GAPDH rabbit polyclonal antibody (1.0 µg for 1.0 mg of total protein lysate, Proteintech, Cat No. 60004-1-Ig).
Animal experiments
BALB/c nude mice were purchased from GemPharmatech Experimental Animal Co., Ltd (Nanjing, China) and maintained in a specific pathogen-free (SPF) animal area. These experiments were approved and supervised by the Experimental Animal Ethics Committee of Ruiye model animal (Guangzhou) Biotechnology Co., Ltd (Lunshen [2020] No. 2020010). In the subcutaneous tumor growth xenograft models, 1 × 106 SW480 cells were mixed with 50 ml of Matrigel, then the mixture was injected into subcutaneous tissues. On day 14 after introduction, the subjects were treated with DMSO, 2 mg/kg, and 4 mg/kg of WA every 3 days via intraperitoneal (IP) administration [14]. Meanwhile, the body mass and tumor volume were measured. The mice were sacrificed and the tumors were collected for immunohistochemical staining.
To establish the in-vivo metastasis model, HIF-1α gene knock-down stable SW480 CRC cells (2 × 106 cells/mice) or shNC control cells (2 × 106 cells/mice) were injected venously into the tail. Seven weeks after the injection, all these mice were sacrificed and lung metastatic lesions were removed and examined by IHC and HE staining.
RNA sequencing and screening of differentially expressed genes (DEGs)
To investigate the molecular mechanism of action, RNA sequencing was used to examine differential gene expression. HCT116 and SW480 cells were treated with 0 and 0.2 mM WA, respectively, for 12 h. The Nanodrop 2000 was used to detect total RNA. Hybridization was performed by Origingene Bio-pharm Technology (Shanghai, China). RNA sequencing was as described in the literature [15]. The data set was normalized using the expectation-maximization algorithm available in RNA-Seq. In this study, the analysis of the DEGs was as described in the literature [1].
Statistical analysis
All data were expressed as mean ± standard deviation, and p < 0.05 was considered statistically significant. The tumor volume was determined using repeated measurements of the general linear model and log-rank test with SPSS 18.0. Other statistical analyses used in this research include one-way analysis of variance (ANOVA) and Student’s t-test.
Results
WA inhibits the growth, invasion, and migration of CRC cells in vitro
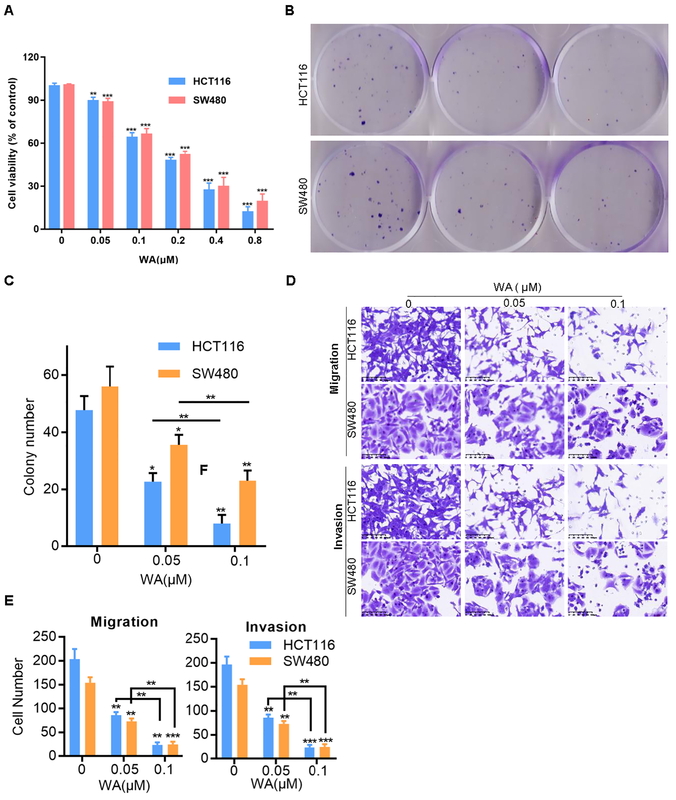
The chemical structure of WA is depicted in Supplementary Figure S1. To examine the anticancer effect of WA on CRC cells, we first used different concentrations of WA to treat human CRC cells to detect its cytotoxicity. The results showed that WA could significantly inhibit CRC cells (Figure 1 A). We also evaluated the effect of WA on CRC cells’ colony formation. Moreover, the data showed that WA significantly reduced the colonies of HCT116 and SW480 cell lines (Figures 1 B, C). Additionally, WA brought about a significant inhibitory effect on CRC cell migration and invasion, as seen in our migration and invasion assays (Figures 1 D, E). In summary, these findings led us to believe that WA exerted a significant inhibitory effect on the growth, migration, and invasion of CRC cells in vitro.
Figure 1
WA inhibits the growth, invasion and migration of CRC in vitro. A – CRC cells were treated with specified concentrations of WA for 24 h and then CCK8 was used for the cell viability analysis. The data were obtained from three independent experiments. B – HCT116 and SW480 cells were treated with specified concentrations (0, 0.05 and 0.1 μM) of WA for 12 days. The colony was fixed and then stained. C – Data come from three independent experiments (B), *p < 0.05, **p < 0.01, ***p < 0.001. D – Migration and invasion assays were used to detect the influence of WA on the migration and invasion ability of CRC cells, respectively. E – Data come from three independent experiments. These experiments were performed three times independently, *p < 0.05, **p < 0.01, ***p < 0.001
WA inhibits the growth and metastasis of CRC cells in vivo
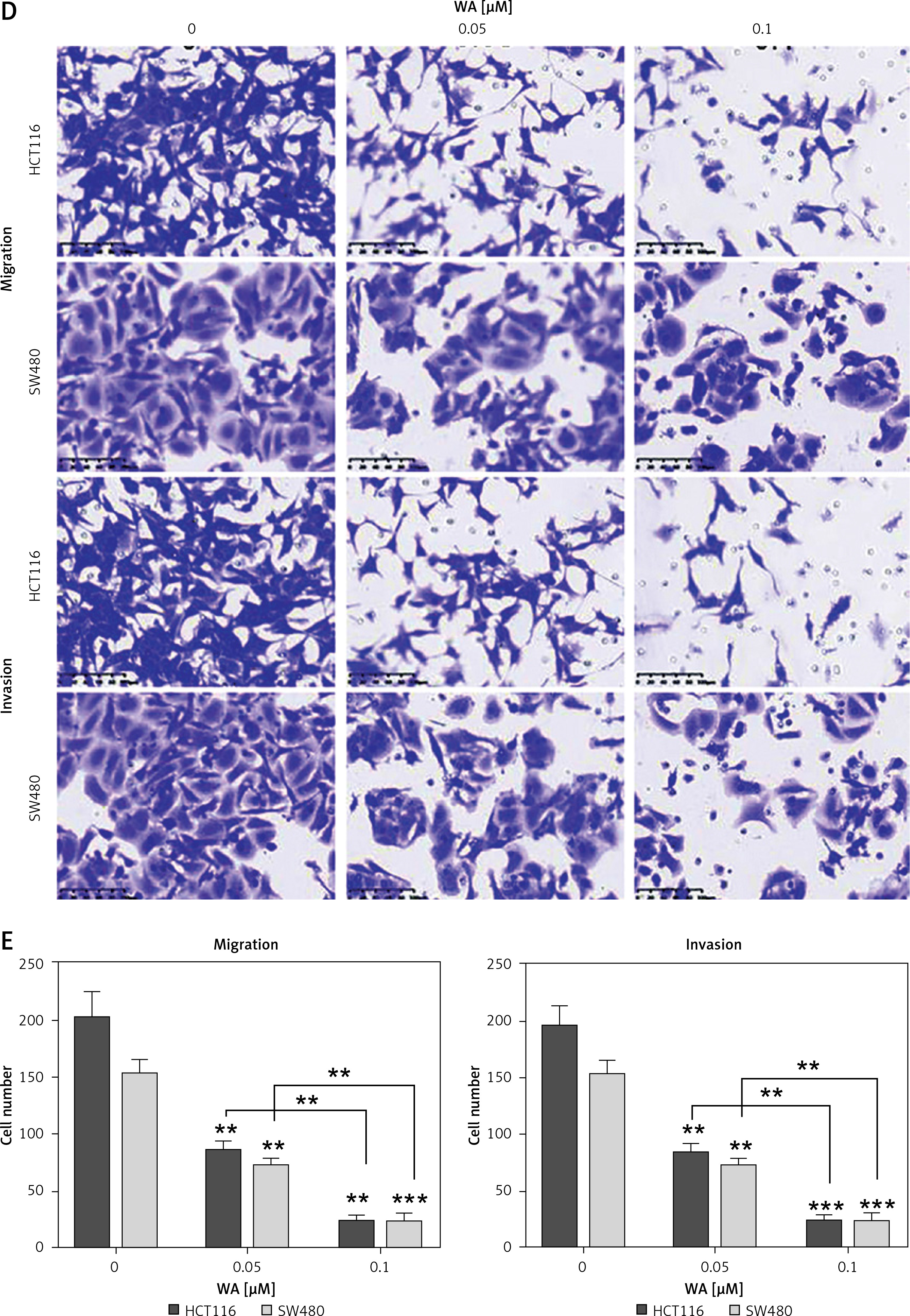
We further verified the effect of WA on the CDX model of CRC and found that WA can inhibit growth in vivo (Figures 2 A–C). In addition, WA has a good inhibitory effect on the metastasis of CRC cells in the metastatic tumor model in which SW480 cells were injected venously into the tail of nude mice (Figures 2 D–F). There was no significant difference in weight loss in the mice during treatment (Supplementary Figure S2 A), and we did not observe a significant difference in serum AST and ALT levels either (Supplementary Figure S2 B). Our last point of interest – liver, kidney, or heart function – yielded no negative impact (Supplementary Figure S2 C). Taken together, the results indicated that WA not only significantly inhibited the growth and metastasis of CRC cells in vivo, but also maintained a low level of cell toxicity. The mechanism of WA lowering the growth, migration, and invasion of CRC cells is unknown; hence, we were incentivized to conduct further experiments.
Figure 2
WA inhibits the growth and metastasis of CRC in vivo. A–C – SW480 cells were subcutaneously inoculated into nude mice. The mice were randomly divided into 3 groups (n = 6), and intraperitoneal injections of DMSO, WA 2 mg/kg, WA 4 mg/kg were injected into different groups daily for 30 days. A – Tumor volume measurement. B – Subcutaneous tumor tissue of the 3 groups. C – Tumor mass measurement. The scale bar is shown in the figure. D–F – Lung metastasis model after injection of SW480 cells via tail vein. They were randomly divided into 3 groups (n = 6), and intraperitoneal injections of DMSO, WA 2 mg/kg, WA 4 mg/kg were administered to different groups daily for 30 days. D – Representative images of metastatic lung tumors. E – Quantification of metastatic lung tumors. F – Measurement of the weight of the whole lung. Scale bar, 1 cm. *p < 0.05, **p < 0.01, ***p < 0.001
WA inhibits the EMT pathway and its genes in CRC cells
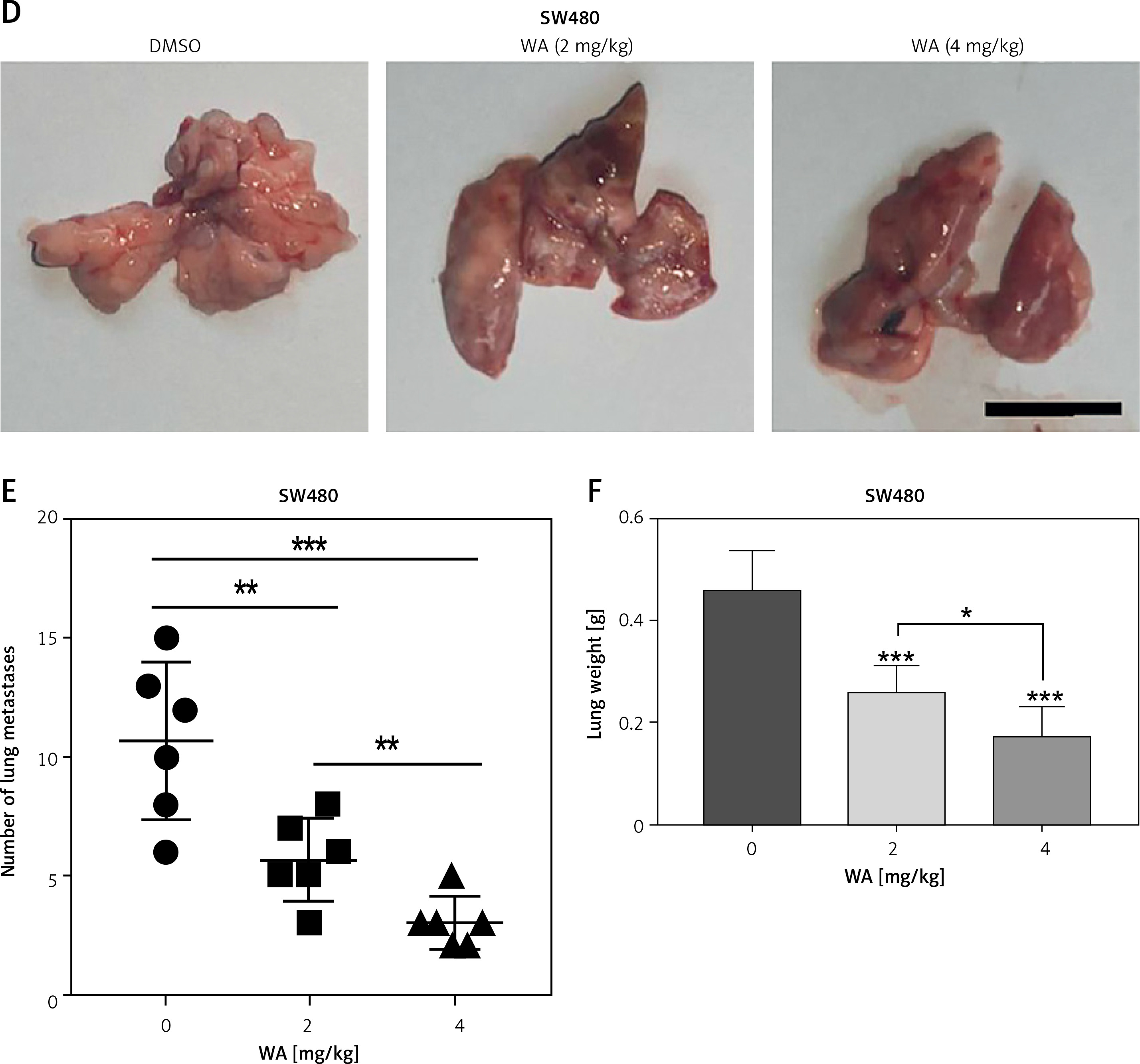
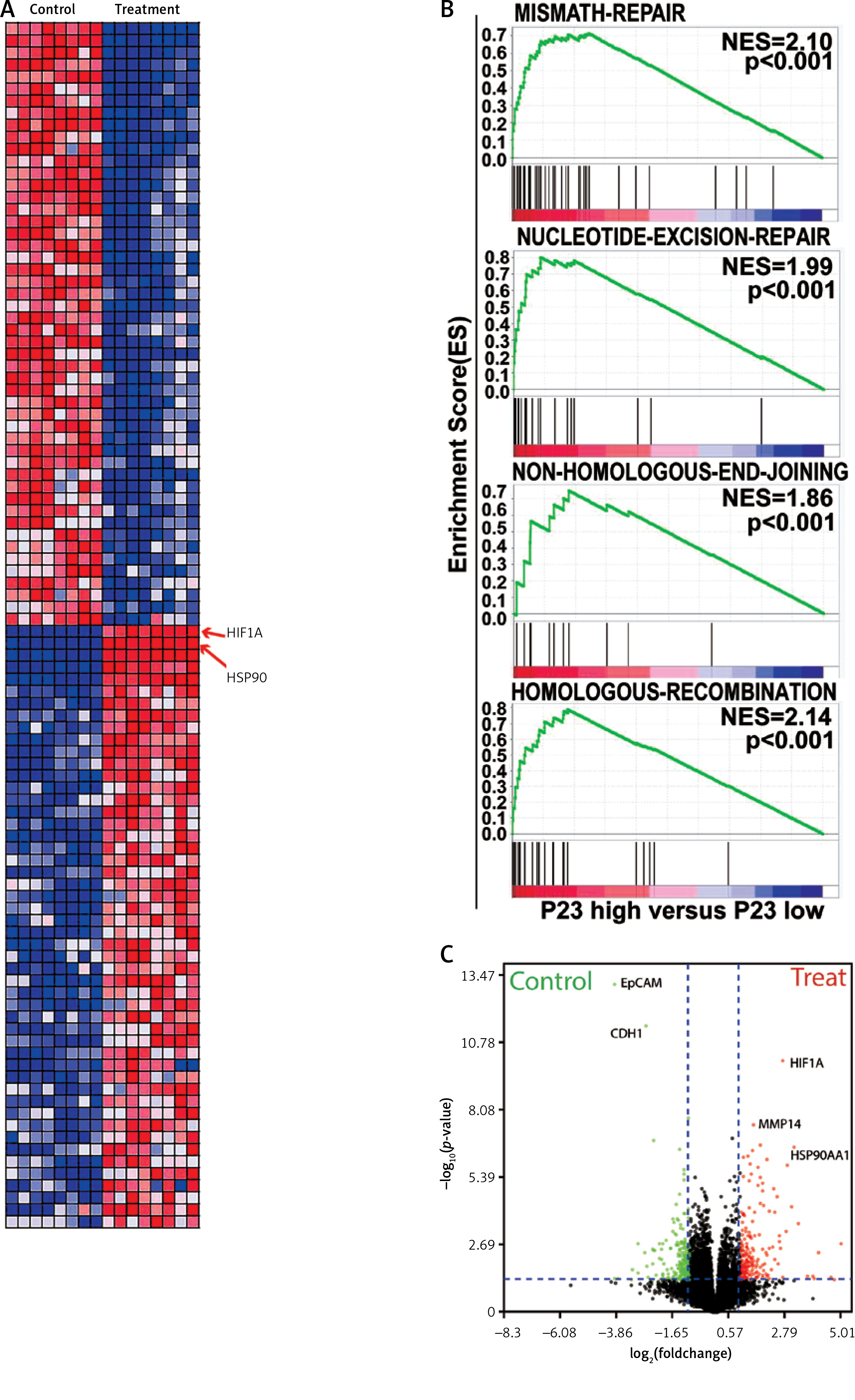
To explore the mechanism deployed by WA that interferes with the growth and metastasis of CRC, we sequenced the mRNA of CRC cells treated with DMSO and WA. The data were analyzed by constructing a protein-protein interaction (PPI) network. Figure 3 A displays their DEGs as well as fold change. Pathway enrichment analysis shows that WA mainly inhibits the EMT-related pathways (Figure 3 B); DEGs show that WA has a good inhibitory effect on HIF-1α, HSP90, and other genes (Figure 3 C). We know that HIF-1α is an important gene in the EMT pathway and also a client protein of HSP90, with HSP90 stabilizing HIF-1α. Our results suggest that WA may inhibit the growth and metastasis of CRC cells by suppressing the HSP90/HIF-1α/EMT axis in CRC.
Figure 3
WA inhibits the EMT pathway and its genes in CRC. A – Heatmap of mRNA differential expression of HCT116-c and cSW480-c cells against HCT116-e and SW480-e cells. Red represents high and blue represents low. The abscissa represents the gene name. B – Pathway enrichment was performed on CRC cells treated with DMSO and WA. C – DEGs of the transcriptome sequencing
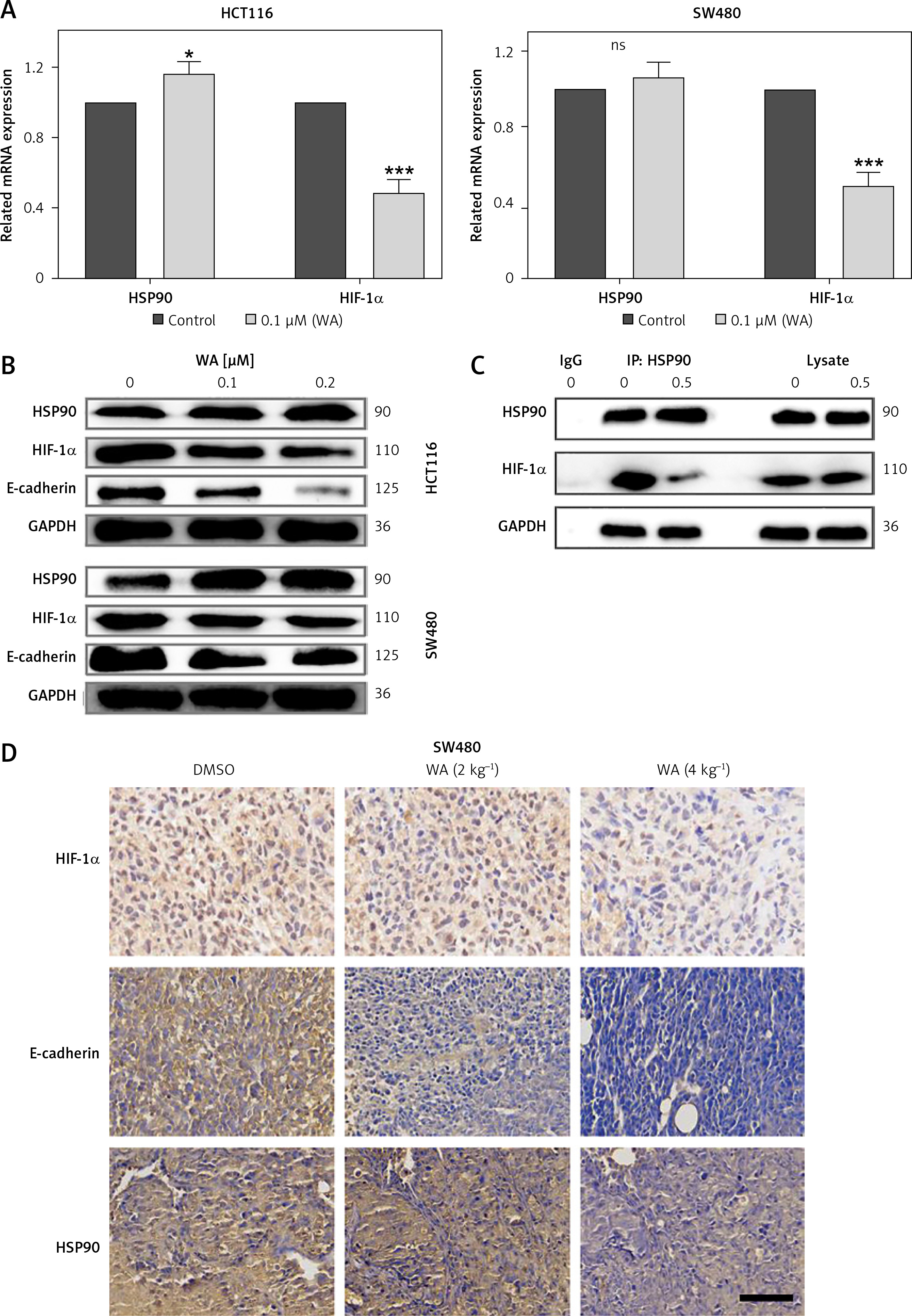
WA inhibits HSP90/HIF-1α in CRC cells by EMT
Our results proved that WA can inhibit HSP90, HIF-1α, and their interactions in CRC by EMT, but we still need to explore the role of HIF-1α in CRC. To verify WA’s ability to inhibit the HSP90/HIF-1α/EMT axis, we performed q-PCR detection. The results showed that WA can effectively inhibit the expression of HIF-1α mRNA in CRC (Figure 4 A). Western blot revealed that WA can effectively inhibit the expression levels of Hsp90, HIF-1α and E-cadherin (Figure 4 B). The results of Co-IP indicate that WA can effectively attenuate the interaction between Hsp90 and HIF-1α (Figure 4 C). IHC also confirmed that WA can strongly inhibit HSP90, HIF-1α and E-cadherin (Figure 4 D).
Figure 4
WA inhibits HSP90/HIF-1α pathway of CRC. A – Treatment of CRC cells with WA at the specified concentration for 12 h, and qPCR detection of related mRNA expression levels. B – After treating CRC cells with DMSO or WA at the specified concentration for 24 h, western blotting analysis was used to detect the expression level of proteins, including HSP90, HIF-1α, and E-cadherin. C – SW480 cells were treated with 1 μM WA or without 1 μM AIL for 24 h. They were then treated with HSP90 antibody IP, and the specified antibody was used to detect the co-immunoprecipitated protein. D – Representative images of immunohistochemical detection of HSP90, HIF-1α, and E-cadherin in CDX tumor tissue (400×). Scale bars, 50 μm. These experiments were performed three times independently, *p < 0.05, **p < 0.01, ***p < 0.001
Our previous research demonstrated that WA can inhibit EMT (epithelial-mesenchymal transition). We have now discovered that WA can function by inhibiting HSP90/HIF-1α. It raises the question of whether WA’s inhibition of HSP90/HIF-1α is responsible for its inhibitory effect on EMT.
To investigate this, we used 17-AAG, a classic inhibitor of HSP90, on SW480 and HCT116 cells and detected the levels of epithelial marker CDH1 (E-cadherin) and mesenchymal marker Vim (vimentin) via RT-qPCR. We found that inhibition of HSP90 led to a significant increase in CDH1 and a significant decrease in Vim (Supplementary Figures S3 A and S4 A). Conversely, overexpression of HSP90 resulted in a significant decrease in CDH1 and a significant increase in Vim (Supplementary Figures S3 B and S4 B). When WA was reintroduced into HSP90 overexpressing cells, CDH1 increased significantly while Vim decreased significantly (Figures S3C and S4C). This indicates that WA exerts its EMT inhibitory effect through the inhibition of HSP90.
Furthermore, when we inhibited HSP90 and simultaneously overexpressed HIF-1α, we observed a significant decrease in CDH1 and a significant increase in Vim (Supplementary Figures S3 D and S4 D). This suggests that the effect of HSP90 is dependent on HIF-1α.
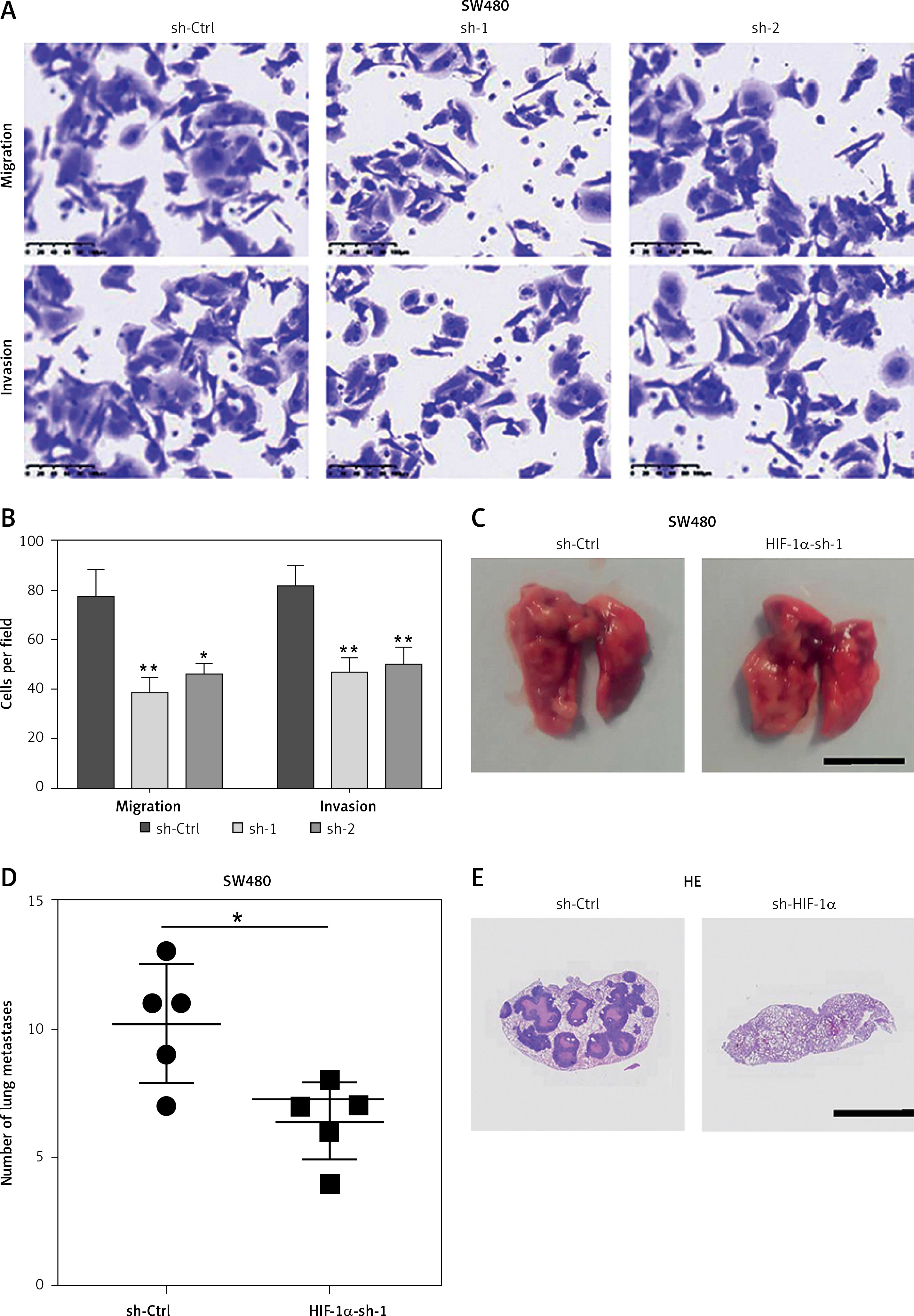
HIF-1α is an important gene that promotes CRC metastasis
We performed migration and invasion experiments on cell lines with HIF-1α knockdown, and the outcome showed that the migration and invasion ability of these CRC cells were weakened (Figures 5 A, B). The results of the lung metastasis model with HIF-1α knockdown SW480 in nude mice are consistent with those in vitro (Figures 5 C–E). These results indicate that HIF-1α is an important gene that promotes CRC metastasis.
HSP90 stabilizes and regulates HIF-1α
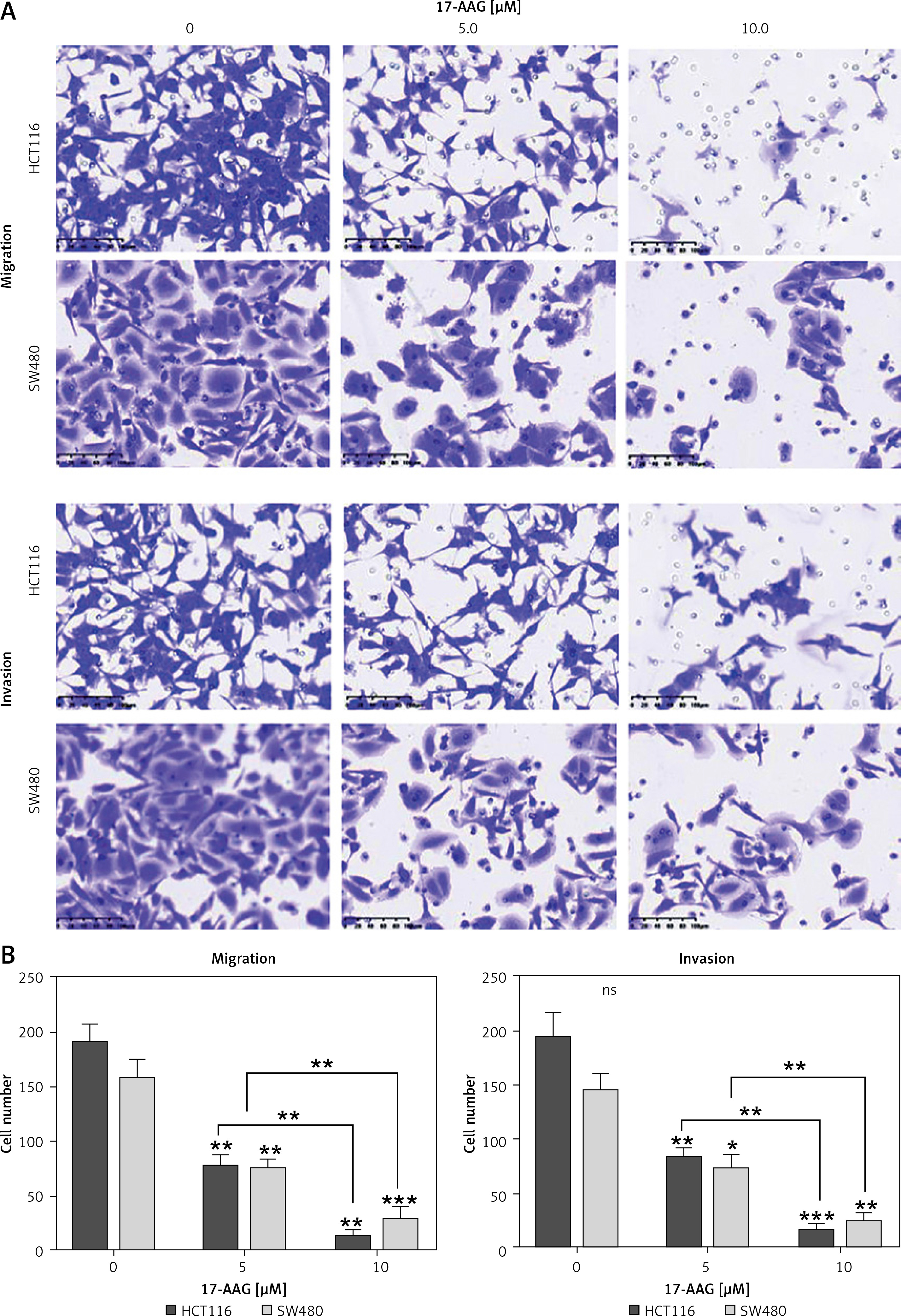
To explore whether HSP90 has a regulatory relationship with HIF-1α, we introduced 17-AAG, a classic inhibitor of HSP90. We found that 17-AAG can shut down the migration and invasion of these CRC cells (Figure 6 A). 17-AAG can inhibit the HIF-1α mRNA level (Figure 6 B) and protein expression level (Figure 6 C) of CRC. These results indicate that HSP90 can stabilize and regulate HIF-1α.
Figure 6
HSP90 stabilizes and regulates HIF-1α, and HIF-1α promotes metastasis of CRC cells. A – Migration and invasion assays to detect the effects of different concentrations of 17-AAG on the migration and invasion ability of HCT116 and SW480 cells. B – Data come from three repeated experiments (A), *p < 0.05, **p < 0.01, ***p < 0.001. C – HCT116 and SW480 cells were treated with the specified concentration of 17-AAG for 12 h, and the mRNA expression level of HIF-1α was detected by qPCR. D – After treating HCT116 and SW480 cells with DMSO or WA at the specified concentration for 24 h, western blotting was used to detect the expression levels of HSP90 and HIF-1α proteins. These experiments were performed three times independently, *p < 0.05, **p < 0.01, ***p < 0.001
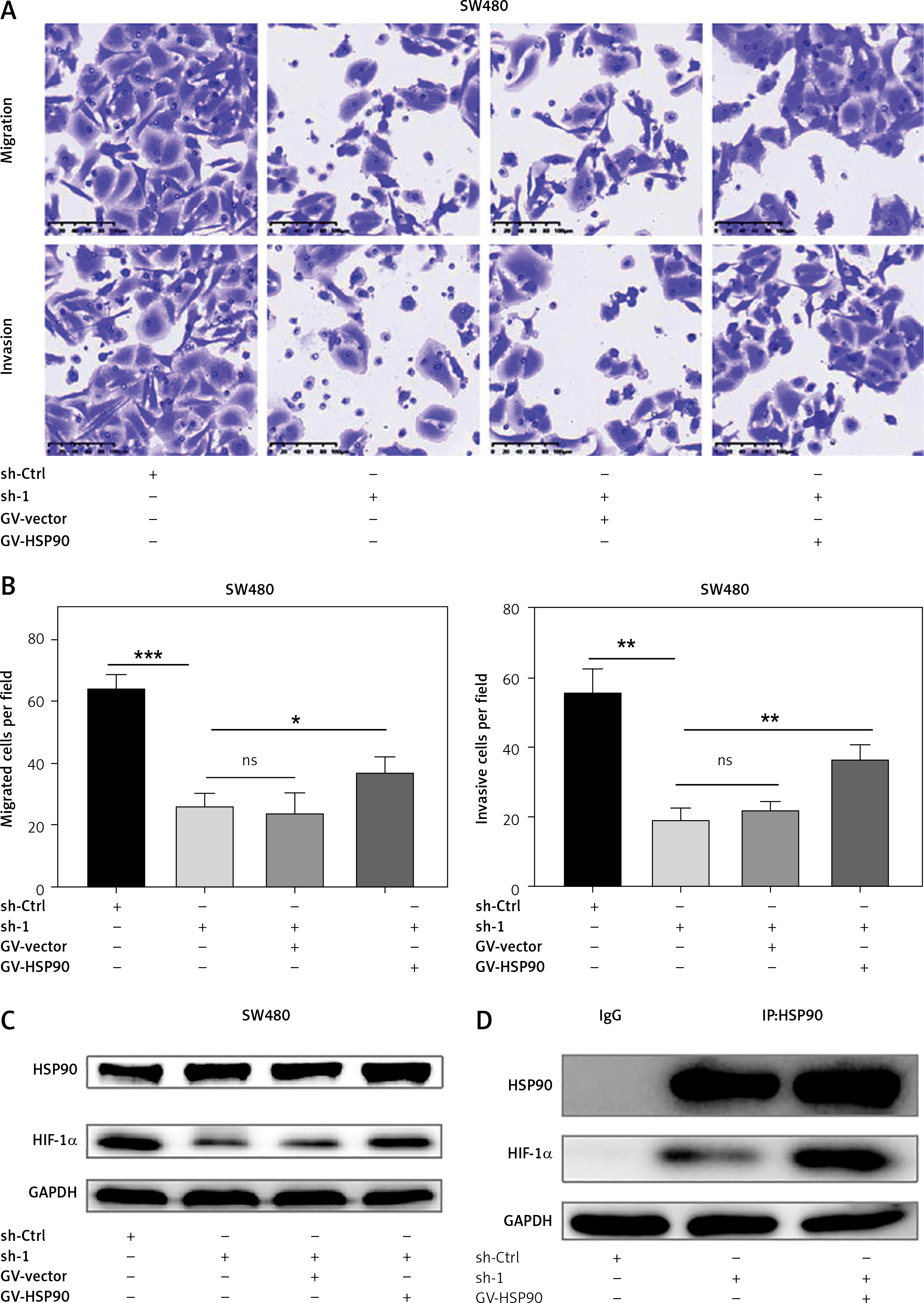
HSP90/HIF-1α axis promotes CRC cell migration and invasion
Although HIF-1α plays an important role in promoting CRC metastasis, it is unclear whether the HSP90/HIF-1α axis is involved in CRC metastasis at all. As mentioned above, we have established stable HIF-1α knockdown and control SW480 cells, and also established a HSP90 (GV-HSP90) overexpression model and control plasmid (GV-Vector) model in these cell lines. Transwell experiments showed that the decline in migration and invasion of HIF-1α knockdown SW480 cells can be reversed by the overexpression of HSP90 in the same cells (Figures 7 A, B). It was confirmed by western blotting and Co-IP that overexpression of HSP90, HIF-1α, and HSP90-HIF-1α in HIF-1α knockdown SW480 cells caused it to be successfully rescued (Figures 7 C, D). In summary, the HSP90/HIF-1α axis can promote CRC metastasis.
Figure 7
HSP90/HIF-1α signal axis promotes migration and invasion of CRC cells. A – Migration and invasion ability of sh-Ctrl, HIF-1α-sh1, HIF-1α-sh1 + GV-Vector and HIF-1α-sh1 + GV-HSP90 in the SW480 cells. B – Data come from three independent experiments (A). C – Western blotting was used to detect the HIF-1α protein expression levels of sh-Ctrl, HIF-1α-sh1, HIF-1α-sh1 + GV-Vector and HIF-1α-sh1 + GV-HSP90. D – Co-IP detects expression level of HIF-1α in HSP90 knockdown HIF-1α-sh1, HIF-1α-sh1+GV-HSP90. These experiments were performed three times independently, *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
Although a large number of molecular pathways and markers has been continuously reported over the years, the mortality rate has remained high [16]. HIF-1α as an important transcription factor during hypoxic stress is seen in glycolysis, angiogenesis, and cancer metastasis. It is an important protein that promotes tumor metastasis as well [7]. Targeting HIF-1α may block many key pathways of tumor growth.
Natural products are an important source for identifying new anti-cancer drugs. To date, several clinically used antitumor drugs have been derived from plant, such as taxanes, a chemotherapeutic agent treating ovarian cancer, breast cancer and prostate cancer. WA, an extract of Withania somnifera, has shown antidiabetic, anti-inflammatory, anti-cancer, and anti-angiogenic effects in several studies [17–19]. Through our research on WA in CRC, we found that WA effectively inhibits the growth and metastasis of CRC in vitro and in vivo (Figures 1 and 2) and has low toxicity (Supplementary Figure 2). Further transcriptome analysis suggests that WA inhibits multiple EMT pathways of CRC and targets HIF-1α and HSP90 (Figure 3). HSP90 not only acts as a molecular chaperone of HIF-1α, forming HSP90-HIF-1α [20], but it also plays a role in stabilizing and regulating HIF-1α [7]. We also confirmed the HIF-1α regulatory effect of HSP90 on CRC through rescue experiments (Figure 5). Heat shock proteins are a group of evolutionarily conservative molecular chaperones that stabilize and activate more than 200 proteins and participate in DNA repair, protein homeostasis, transcription regulation, chromatin remodeling, etc. [21]. HSP90 is an essential player in cancer development, and targeting HSP90 is one of the most viable and important strategies to inhibit cancer growth [22]. HSP90 inhibitors have already been studied in preclinical and clinical trials as new anti-cancer drugs [23]. WA is known to target HSP90 and block the interaction of multiple client proteins such as CDK4, Cyclin-D1, Akt, Raf-1, and CDC37 [24]. In our experiment, we found that WA can inhibit HSP90 very well (Figures 4 B–D). HIF-1α is an important gene in the EMT pathway [2, 25]. WA also inhibits HIF-1α and E-cadherin, the EMT marker protein of CRC.
Figure 5
HIF-1α is an important protein that promotes tumor metastasis. A – Migration and invasion ability of sh-Ctrl, HIF-1α-sh1 and HIF-1α-sh2 in the SW480 cells. B – The data come from three repeated experiments (A), C – Representative images and quantitative analysis of metastatic lung tumors after injection of knockdown HIF- 1α or control group SW480 cells. D – The data come from three repeated experiments (C). E – Representative images of metastatic lung tumors with HE staining. Scale bar, 1 cm. These experiments were performed three times independently, *p < 0.05, **p < 0.01
EMT is an important biological process in tumor cells. After EMT, tumor cells have stronger metastasis and invasion capabilities, participate in the migration of primary cancer cells to secondary sites, and promote metastasis. In addition, EMT has characteristics such as drug resistance, immune evasion, and stemness, which help successful distal metastases [8, 26–30]. In many solid tumors, hypoxia is a prominent feature of the microenvironment niche due to insufficient vascularization. Tumor cells respond to changes in hypoxia through the transcription factor HIF-1α to orchestrate a large number of its basic cellular functions. In recent years, it has been found that EMT has a close relationship with hypoxia, and the activation of the hypoxia-driving gene HIF-1α can promote EMT [31]. Also, tumor hypoxia catalyzes more HIF-1α activation and can enhance the aggressiveness of cancer to a greater extent, including the enrichment of some EMT phenotypes. However, no selective HIF-1α inhibitors have been approved clinically so far. In view of the profound influence of HIF-1α on cancer progression through gene expression, the development of HIF-1 direct or indirect inhibitors and biological research on the HIF-1α pathway have garnered great interest [19, 32, 33].
In conclusion, we found that WA has good anti-cancer potential. It can inhibit the growth and metastasis of CRC by targeting the HSP90/HIF-1α/EMT axis. It can be considered as a new potential drug candidate for CRC treatment.