Introduction
In the complicated process of renal cell carcinoma (RCC) carcinogenesis, along with the vascular endothelial growth factor (VEGF)-tyrosine kinases-m-TOR pathway, host immunosurveillance plays a pivotal role. Different therapies targeting the VEGF pathway have proven but limited efficacy and multiple drugs modulating the immunological system have been introduced [1–3]. In general, the immunologic system should be able to eliminate neoplastically changed cells, but cancer cells have evolved many mechanisms to evade the immunological response, and therefore, in many patients, immunosurveillance is ineffective. In the immunological response, T cell activation plays a key role. This process is controlled by a complicated network of interactions of positive and negative regulators including CD28, which is the primary co-stimulatory molecule, and a variety of co-inhibitory molecules, among others: cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), B-and T-lymphocyte attenuator (BTLA) and programmed cell death protein 1 (PD-1). Under normal conditions, down-regulatory proteins act to limit T-cell responses to chronic antigen exposure, but in cancer patients their function might be altered, which could cause impaired elimination of cancer cells [4, 5].
CD28 is co-stimulatory molecule expressed in CD3+ thymocytes, 95% of CD4+ T cells, γ/δ T cells and approximately 50% of CD8+ T cells. The costimulatory signal provided by this receptor after binding with its ligand B7-1 (CD80) or B7-2 (CD86) presented on antigen presenting cells (APCs) is critical for allowing full T cell activation, sustaining cell proliferation, preventing anergy and/or apoptosis and inducing differentiation to effector and memory status [6]. CTLA-4 is only minimally expressed in resting T cells, and is transiently up-regulated following stimulation [7]. The binding of CTLA-4 with its ligands (which are common to CD28) causes inhibition of downstream T-cell receptor (TCR) signaling by SH2 domain-containing phosphatases, inhibition of lipid-raft and microcluster formation, and negative regulation of the immune response via extrinsic components, such as transforming growth factor-β (TGF-β) and the tryptophan-degrading enzyme indoleamine 2,3-dioxygenase (IDO) [8]. Moreover, CTLA-4 competes with costimulatory CD28 molecules, having more effective ligation to their common B7 ligands. It has been shown in animal models that CTLA-4 blockade leads to enhancement of the immune response rejection of tumors [9, 10]. BTLA is expressed in a broad range of cells responsible for immunosurveillance, including CD4+ and CD8+ T cells, B cells, natural killer (NK) T cells, macrophages and dendritic cells (DCs) [11]. Its activation is triggered through binding to the herpes virus entry mediator (HVEM), a member of the tumor necrosis factor receptor family, and phosphorylation of either Src homology 2 domain-containing protein tyrosine phosphatase (SHP)-1 or SHP-2, which are responsible for inhibitory signaling [12]. Currently, molecules targeting inhibitory proteins such as CTLA-4 (ipilimumab) or PD-1 (nivolumab, pembrolizumab, etc.) have been introduced into treatment of patients with various tumor types, including kidney cancer [13, 14]. BTLA is seen as the next potential target for anticancer intervention [15].
SNPs are substitutions of single nucleotides at specific locations in the DNA strand. This might result in aberrant protein expression and function causing dysregulation of processes for which it is responsible. We have hypothesized that SNPs occurring within genes encoding immune check-point molecules might influence the immune system response to cancer cells. This in turn might lead to diminished patients survival due to cancer. We have previously demonstrated that SNPs in the CTLA-4, BTLA and CD28 genes are associated with the advanced stages of clear cell renal carcinoma (ccRCC) and with more aggressive forms of tumor [16, 17].
Herein, we tested the hypothesis that these polymorphisms also influence survival in ccRCC patients, as has been demonstrated for CTLA-4 gene variations in patients with other cancers [18–22]. For this purpose, we analyzed overall survival in ccRCC patients in relation to the following polymorphisms: in the CTLA-4 gene: c.49A>G (rs231775), g.319C>T (rs5742909), g.*6230G>A (CT60; rs3087243), g.*10223G>T (Jo31; rs11571302); in the CD28 gene: c.17+3T>C (rs3116496), and CD28c.-1042G>A (rs3181098); and in the BTLA gene: rs2705511, rs1982809, rs9288952, rs9288953, rs2705535, rs1844089.
Material and methods
Patients
This study was approved by Wroclaw Medical University Bioethics Committee (KB 55/2010). All patients gave informed written consent before study commencement. A total of 282 patients with kidney cancer were prospectively recruited by the Department of Urology and Oncologic Urology at Wroclaw Medical University, a tertiary urologic cancer center. The analysis was performed on 214 clear cell renal carcinoma patients with full data regarding genetic and clinical variables, who had completed follow-up. 56 patients were excluded due to non-clear cell histopathology and 11 due to lack of survival data, one for incomplete genotyping data. All patients were followed up every 3 months, either in person or by teleconsultation, to determine their OS.
Genotyping
DNA from each individual was isolated from venous blood using the QIAamp DNA Blood Mini Kit (Qiagen, Germany) according to the manufacturer’s protocol. The following SNPs were genotyped as described previously [14]: g.319C>T in the promoter region, c.49A>G in exon 1, CT60 and Jo31 in the 3′ untranslated region (UTR) of the CTLA-4 gene, c.17+3T>C in the third intron and c.-1042G>A in the upstream region of the CD28 gene. SNPs in the BTLA gene – rs1844089, rs2705535 and rs9288953 (located in intron 1); rs9288952 (located in exon 4), rs1982809 (located in 3′-UTR) and rs2705511 (located in the intragenic region) – were genotyped using the TaqMan SNP Genotyping Assay as described previously [16].
Statistical analysis
Statistical analysis was performed using Statistica, version 13 (Data Analysis Software System, USA). For all quantitative variables, median, minimum and maximum values were calculated. Qualitative variables were analyzed with descriptive statistics, using absolute and percentage frequency distribution. Overall survival with different clinicopathological variables was calculated using Kaplan-Meier analysis. A log-rank test was used to assess the association between predictor variables and patient survival. A Cox proportional hazards model was used to assess the impact of a range of qualitative and continuous variables on survival. The model building process was carried out using a forward/backward stepwise method, and standard measures of goodness-of-fit (AIC, BIC, R2) were used to evaluate the model. Results were considered statistically significant based on a threshold of p < 0.05.
Results
Patient characteristics are described in Table I.
Table I
Patient and disease characteristics
Influence of clinicopathological factors on overall survival of clear cell renal carcinoma patients
As the first step to test our group we analyzed the influence of established survival prognostic factors. A univariate analysis confirmed that known clinicopathological factors – clinical stage, grade of tumor, presence of necrosis on histopathology, presence of venous thrombus and diameter of the tumor of ≥ 70 mm – were significantly associated with overall survival. The presence of sarcomatoid differentiation in kidney tissue was the only clinicopathological parameter not associated with OS in our analysis (Table II).
Table II
Associations between overall survival (OS) and clinicopathological variables
Influence of genetic variations in CTLA-4, CD28 and BTLA genes on overall survival of clear cell renal carcinoma patients
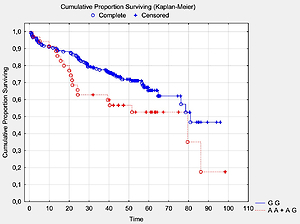
We analyzed the OS in relation to previously genotyped polymorphisms in genes encoding co-signaling molecules CTLA-4, CD28 and BTLA. The analysis of overall survival in relation to the investigated polymorphisms showed that in long term observation the presence of the A allele at the BTLA rs1844089 SNP [AG+AA genotypes] is the only genetic factor significantly negatively correlated with survival in comparison with the GG homozygote (42.5 m-o vs. 48.2 m-o, Table III; Figure 1). For the other SNPs there were no statistically significant associations with OS (Table III).
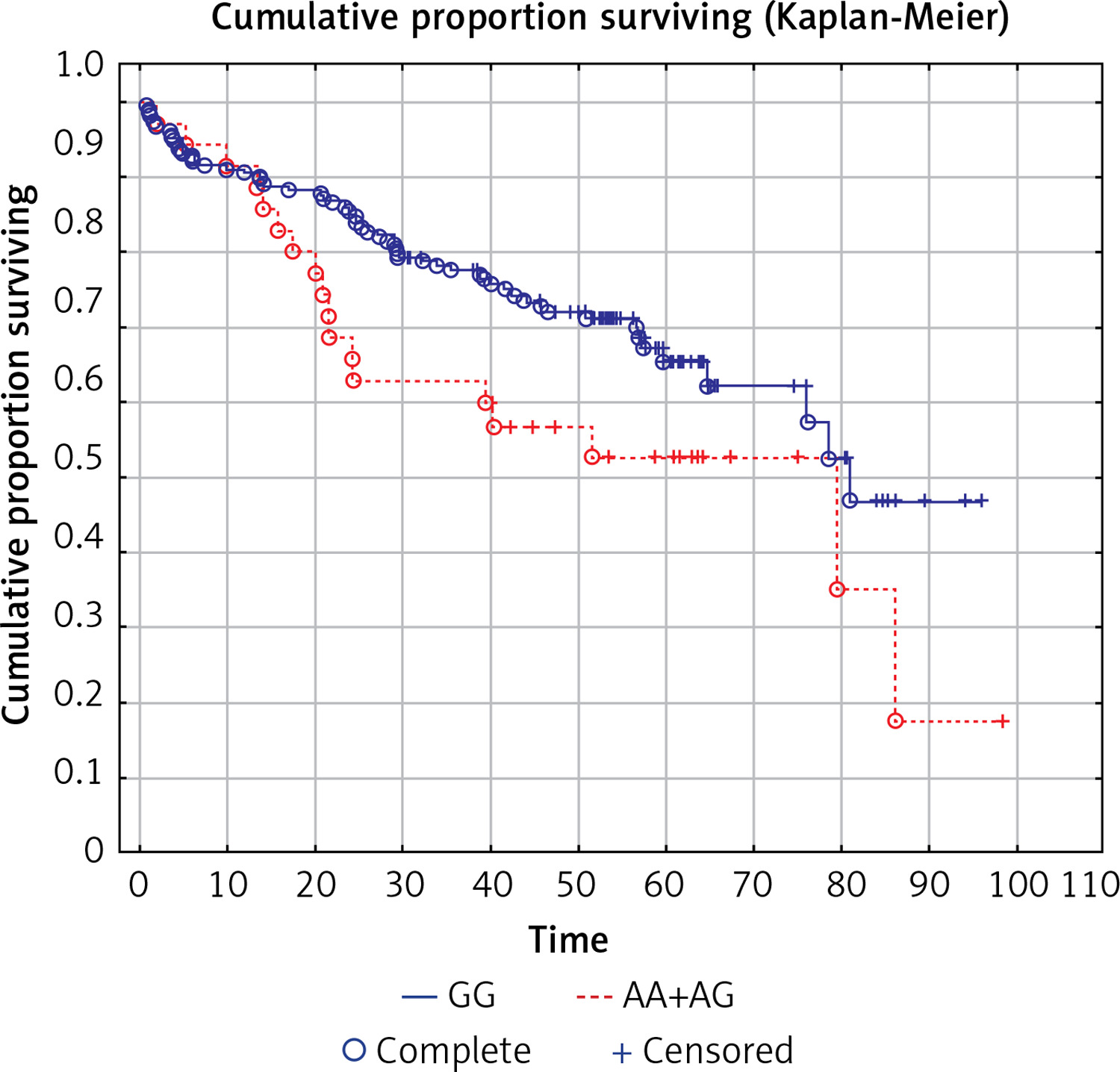
Table III
Analysis of survival by polymorphism (SNPs in the CTLA-4, CD28 and BTLA genes)
| SNP | Genotype | N | Median OS [months] | P-value* |
|---|---|---|---|---|
| CTLA-4c.49A>G (rs231775) | AA | 66 | 43.1 | 0.26 |
| AG | 110 | 48.1 | ||
| GG | 38 | 49.1 | ||
| AA | 66 | 43.1 | 0.30 | |
| AG+GG | 148 | 48.2 | ||
| CTLA-4g.319C>T (rs5742909) | CC | 168 | 46.6 | 0.69 |
| CT | 46 | 48.5 | ||
| CT60G>A (CTLA-4g.*6230G>A; rs3087243) | AA | 23 | 54.4 | 0.18 |
| AG | 115 | 44.5 | ||
| GG | 76 | 48.2 | ||
| GG | 76 | 48.2 | 0.30 | |
| AG+AA | 138 | 45.1 | ||
| Jo31G>T (CTLA-4g.*10223G>T ; rs11571302) | GG | 76 | 51.4 | 0.32 |
| GT | 113 | 44.7 | ||
| TT | 25 | 45.9 | ||
| GT+TT | 138 | 45.1 | 0.15 | |
| GG | 76 | 51.4 | ||
| CD28c.17+3T>C (rs3116496) | CC | 2 | 42.8 | 0.73 |
| CT | 55 | 42.4 | ||
| TT | 157 | 49.0 | ||
| CC+CT | 57 | 42.4 | 0.44 | |
| TT | 157 | 49.0 | ||
| CD28c.-1042G>A (rs3181098) | AA | 16 | 45.3 | 0.36 |
| AG | 93 | 50.9 | ||
| GG | 105 | 45.7 | ||
| GG | 105 | 45.7 | 0.52 | |
| AA+AG | 109 | 50.0 | ||
| BTLA rs1844089 | AA | 3 | 42.5 | 0.12 |
| AG | 32 | 42.5 | ||
| GG | 179 | 48.2 | ||
| AA+AG | 35 | 42.5 | 0.045 | |
| GG | 179 | 48.2 | ||
| BTLA rs2705535 | CC | 206 | 46.1 | 0.99 |
| CT | 7 | 51.6 | ||
| BTLA rs9288952 | AA | 184 | 47.3 | 0.18 |
| AG | 28 | 43.5 | ||
| BTLA rs9288953 | CC | 81 | 51.6 | 0.29 |
| CT | 102 | 47.3 | ||
| TT | 31 | 41.8 | ||
| CC+CT | 183 | 50.0 | 0.23 | |
| TT | 31 | 41.8 | ||
| BTLA rs1982809 | AA | 112 | 47.3 | 0.17 |
| AG | 87 | 46.3 | ||
| GG | 15 | 40.0 | ||
| AA+AG | 199 | 46.6 | 0.18 | |
| GG | 15 | 40.0 | ||
| BTLA rs2705511 | AA | 110 | 45.3 | 0.16 |
| AC | 90 | 49.0 | ||
| CC | 14 | 43.6 | ||
| AA | 110 | 45.3 | 0.09 | |
| AC+CC | 104 | 46.9 |
Multivariate analysis of the influence of clinicopathological factors and genetic variations in CTLA-4, CD28 and BTLA genes on overall survival of clear cell renal carcinoma patients
Various Cox models were created and a retrograde Cox regression model including significant clinical parameters and investigated polymorphisms was selected on the basis of AIC, BIC and R2. We confirmed the results obtained in univariate analysis that the BTLA rs1844089 SNP, and also tumor stage ≥ 3, tumor grade 4 and tumor diameter ≥ 70 mm, were all significantly negatively associated with overall survival (Table IV). In particular, the presence of the A allele at the BTLA rs1844089 SNP [AG+AA genotypes] increased the hazard ratio (HR) of death by more than two-fold. Other parameters – necrosis, presence of venous thrombus, aggressive phenotype – were not significantly associated in multiparametric analysis.
Table IV
Results of retrograde Cox regression analysis of the influence of variables on probability of death
In silico analysis of functional role of rs1844089 polymorphism
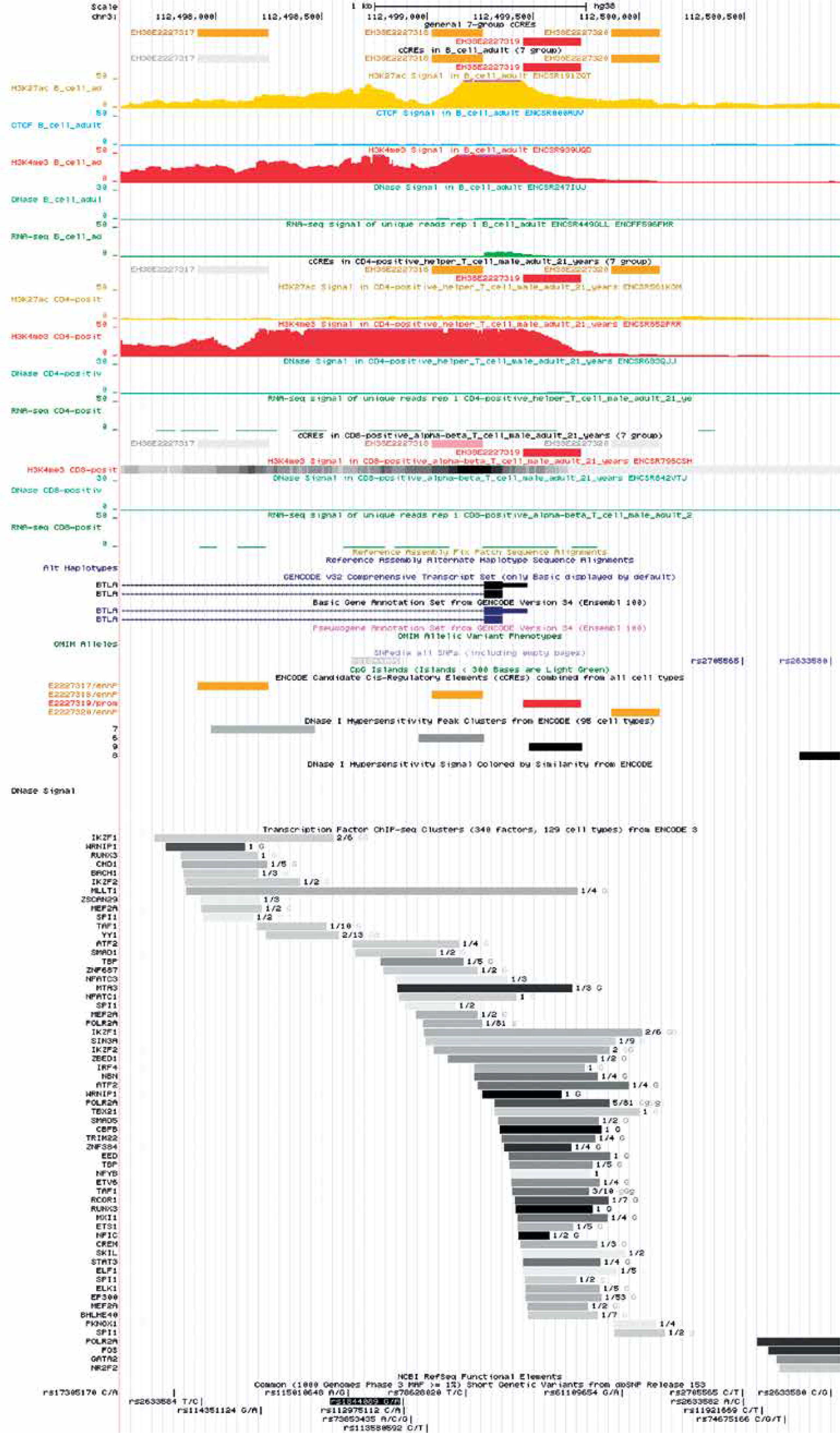
In order to evaluate the potential biological relevance of rs1844089 we performed an in silico analysis with application of publicly available tools. According to the Encyclopedia of DNA Elements (ENCODE) this genetic variant is located in close proximity (565 kb) to the candidate cis-regulatory element (cCRE) EH38E2227319, which has been predicted to have a promoter-like signature in B cells, and as well as GM12878 and OCI-LYF (human B cell non-Hodgkin lymphoma) cell lines.
Moreover, on the basis on in silico analysis using ENDCODE and RegulomeDB (Figure 2) it can be predicted that metastasis-associated protein 3 (MTA3) interacts with the BTLA gene within the region where the rs1844089 SNP is situated (Figure 2).
Figure 2
Location of rs1844089 of BTLA gene in relation to data from ENCODE project showing potential regulatory element-binding sites upstream and downstream of rs1844089 in B cells, CD4+, and CD8+ cells. The figure was downloaded from the UCSC genome browser (http://genome.ucsc.edu). A view of the region surrounding the rs1844089 also includes the representation of the ChIP-seq data from the ENCODE project
Discussion
The CTLA-4, BTLA and CD28 molecules are pivotal co-signaling proteins regulating lymphocyte activation. Therefore, SNPs in genes encoding these receptors have previously been investigated as potential risk factors in both oncological and non-oncological disease [16–18, 23–28]. Our previous study showed that polymorphisms in the CTLA-4, CD28 and BTLA genes confer susceptibility to RCC. In particular, we found that the presence of a G allele at SNPs CT60 and Jo31 was associated with a higher risk of advanced stage and necrosis of tumor in ccRCC patients [16, 17], particularly in patients heterozygous at both CT60AG and Jo31GT [17]. Women carrying the CD28c.-1042G>A[GG] genotype were more prone to ccRCC [17]. Additionally, we found that the presence of the BTLA rs1982809[GG] genotype was associated with increased risk of RCC and with high grade tumors in ccRCC patients [16].
In this study, we investigated an unselected population of ccRCC patients treated in a tertiary cancer center. This approach allows our results to be generalized for all patients treated due to ccRCC. We analyzed SNPs in the co-signaling molecule genes CTLA-4, BTLA and CD28 as possible factors associated with overall survival in ccRCC patients. Firstly, we investigated whether our population was representative of a typical kidney cancer population. We observed that all classical factors commonly associated with poor prognosis [29] are also present in our study population (Table II), the only exception being sarcomatoid differentiation in cancer tissue, which in our group was not significantly associated with poor prognosis. The potential reason for this observation is the low number of patients with sarcomatoid differentiation (n = 9). We also checked whether commonly accepted tumor aggressive phenotype features (presence of at least one of the following: sarcomatoid differentiation, presence of necrosis, grade 3 or 4, presence of venous invasion) are associated with survival, and the same for tumor diameter in a dichotomous model (< 70 mm or ≥ 70 mm).
Then, we analyzed the associations between polymorphisms in co-signaling molecule genes and prognosis in ccRCC patients. Herein, for the first time, we observed that variation within the BTLA gene influences overall survival in ccRCC patients. We found that the presence of the A allele at the BTLA rs1844089 SNP (AG + AA genotypes) was independently associated with risk of death in a multivariate Cox model. Moreover, we found that presence of the A allele at the BTLA rs1844089 SNP (AG + AA genotypes) was associated with decreased OS in our group by nearly 6 months.
SNPs situated in regulatory regions such as promoters, introns and 3′ UTR regions can play an important role in phenotype. For example, SNPs located in promoter regions may influence promoter activity and mRNA expression level of a gene, and polymorphisms located in the 3′ UTR region might be situated within transcriptional enhancer or microRNA binding sites. It has been reported that the first intron within most genes is important for the splicing process and may regulate gene expression more efficiently than other introns [30].
The BTLA SNP rs1844089 is located in intron 1 of the BTLA gene, and it is postulated that this SNP might be associated with aberrant splicing, which could influence the function of the BTLA protein [31]. This SNP has also been associated with disease risk, tumor size, estrogen and progesterone receptor expression, and C-erbB and P53 status in breast cancer patients [32]. Ge et al. also reported that this SNP was associated with colon cancer risk in Chinese patients, but that the influence of this polymorphism was modified by pork consumption [24]. We investigated BTLA rs1844089 as a risk factor for RCC and ccRCC development, but did not find a statistically significant correlation [16].
As we concluded on the basis of in silico analysis, it can be postulated that this polymorphism is situated in close proximity to cCRE, which may have promoter activity in B cells, where the BTLA expression level is highest. Moreover, it can be predicted that metastasis-associated protein 3 (MTA3) interacts with the BTLA gene within the region where the rs1844089 SNP is situated.
MTA3 belongs to the metastasis-associated protein (MTA) family, all members of which serve as subunits of nucleosome remodeling and the deacetylase (NuRD) protein complex. MTAs play a role in chromatin remodeling and modifying gene expression through interactions with histone- or non-histone proteins and modified nucleosomes while recognizing specific post-translational modification. Although the MTA proteins do not directly bind with DNA, they interact with a variety of chromatin remodeling factors and complexes with enzymatic activities and thereby lead to the repression or derepression of target genes [33].
An emerging body of evidence indicates that MTA3 could regulate cell proliferation and differentiation in various cancers. However, it can act differently in various cancers. It acts as an oncogene in breast cancer and non-small cell lung cancer (NSCLC), and its level is increased in the progression of the above-mentioned tumors [34, 35], while MTA3 expression was significantly reduced in both tumor tissues and cell lines in gastroesophageal junction adenocarcinoma [36] as well as in patients with endometrioid adenocarcinoma [37]. In the study by Ding et al. [38] significantly lower expression of MTA3 in tumor ccRCC tissues compared to normal tissue was noted. What is more, the authors reported that overexpression of MTA3 suppressed the proliferation and migration of ccRCC, indicating that MTA3 played a key role in the pathophysiologic process of ccRCC. One can speculate that low expression of MTA3 in ccRCC influences MTA3 interaction with the BTLA gene, which in turns disturbs gene translation. The allelic variation within the rs1844089 SNP could influence the strength of MTA3 interaction.
The functional role of this polymorphism requires further elucidation, and exploration as a possible marker for increased risk of shortened survival, which could warrant a personalized approach to treatment. Furthermore, it might differentiate patient responses to future BTLA directed therapy approaches.
On the other hand, in our previous study we found a strong association between rs1982809A>G and ccRCC risk, whereas this SNP was not associated with disease survival. Rs1982809A>G is situated in the 3′UTR nearby gene region of BTLA (73 bp). A search in SNPinfo and FastSNP databases did not provide data regarding potential biological implication of this SNP. However, we speculate that since the 3′UTR region of the gene is the place where epigenetic regulation by microRNA binding takes place, presence of allele variants in this region can modulate miR binding. In fact, the miR511-3p potentially binds to the region where rs1982809 is situated (based on the TargetScan analysis). This hypothesis needs to be verified in an experimental study.
Since these two SNPs regulate gene and protein expression by different mechanisms, they might influence different stages of immune surveillance, and therefore have different impacts on disease risk and clinical outcome.
For the other SNPs included in our analysis, we detected no statistically significant associations with OS. Despite that, it is noteworthy that patients with heterozygous CT60[AG] or Jo31[GT] genotypes lived approximately 10 months shorter than those with CT60[GG] or Jo31[GG] genotypes.
Our study is limited by being single-centered, and as such our treatment results are representative of those of a tertiary cancer center. In our population, there is overrepresentation of stage IV patients, which is also characteristic of tertiary cancer centers. From the initial pool of 282 recruited patients, 67 (23.8%) were excluded as a result of the prospective nature of the study (the histopathology is usually unknown before surgery) and/or a lack of adequate data (3.9%). This leads to the study group being smaller, although the prospective nature of the study allows for the elimination of retrospective study biases. The elimination of even small numbers of patients with inadequate survival data, which is the primary end measure, gives the calculations more credibility than attempts to fill these data gaps with statistical approximations.
In conclusion, our prospective cohort provides a unique opportunity to study the influence of SNPs of three genes responsible for lymphocyte activation in a kidney cancer population. The obtained observations give new insights into the influence of the immunological system on the survival of cancer patients and might be of importance in personalizing patient approaches and designing new drugs. Our results require confirmation in larger studies and in different populations across the globe.