Current issue
Archive
Manuscripts accepted
About the Journal
Editorial office
Editorial board
Section Editors
Abstracting and indexing
Subscription
Contact
Ethical standards and procedures
Most read articles
Instructions for authors
Article Processing Charge (APC)
Regulations of paying article processing charge (APC)
ORTHOPEDICS AND TRAUMATOLOGY / RESEARCH PAPER
Unveiling the Immune Related ceRNA Network in Osteoarthritis: Key Biomarkers and Therapeutic Targets
1
Department of Orthopedic, Huashan Hosiptal Affiliated to Fudan University, China
Submission date: 2024-06-14
Final revision date: 2024-09-09
Acceptance date: 2024-10-24
Online publication date: 2024-11-02
Corresponding author
KEYWORDS
OsteoarthritisLong non-coding RNACompetitive endogenous RNAImmune cell infiltrationBiomarkersTherapeutic targets
TOPICS
ABSTRACT
Introduction:
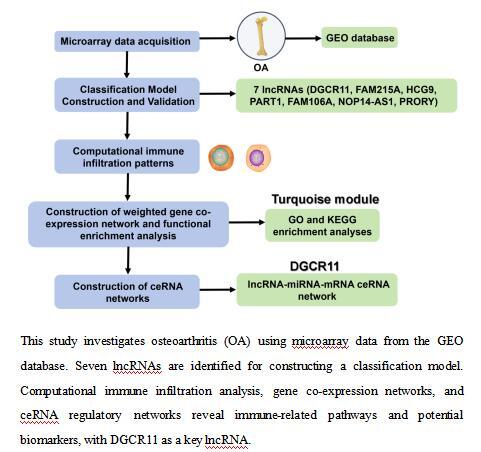
The aim of this study was to explore the immune-related competitive endogenous RNA (ceRNA) network in osteoarthritis (OA), focusing on identifying differentially expressed long non-coding RNAs (lncRNAs), constructing a classification model, and uncovering the associations between these lncRNAs and immune cell subsets in OA.
Material and methods:
Microarray data from the Gene Expression Omnibus (GEO) database was used to identify differentially expressed genes (DEGs) in synovial tissue of OA. A classification model was constructed using the Least Absolute Shrinkage and Selection Operator (LASSO) regression with selected lncRNAs. Computational methods like CIBERSORT were employed to quantify immune cell infiltration patterns, and weighted gene co-expression network analysis (WGCNA) was conducted to delineate co-expression modules. Finally, a ceRNA network was constructed to elucidate the regulatory interactions among lncRNAs, miRNAs, and mRNAs.
Results:
We identified 5927 DEGs, among which 47 were differentially expressed long non-coding RNAs (DELs). Seven DELs (DGCR11, FAM215A, HCG9, PART1, FAM106A, NOP14-AS1, PRORY) formed the basis of a classification model with an area under the receiver operating characteristic (ROC) curve of 1. We also examined the infiltration patterns of immune cells in OA tissues and found significant differences compared to healthy controls, indicating a strong immunological component in OA pathogenesis. WGCNA identified a turquoise module with a high correlation to OA traits.
Conclusions:
The study highlights the importance of ceRNA networks in understanding the complex pathogenesis of OA and offers a comprehensive framework for future research into potential diagnostic biomarkers and therapeutic targets.
The aim of this study was to explore the immune-related competitive endogenous RNA (ceRNA) network in osteoarthritis (OA), focusing on identifying differentially expressed long non-coding RNAs (lncRNAs), constructing a classification model, and uncovering the associations between these lncRNAs and immune cell subsets in OA.
Material and methods:
Microarray data from the Gene Expression Omnibus (GEO) database was used to identify differentially expressed genes (DEGs) in synovial tissue of OA. A classification model was constructed using the Least Absolute Shrinkage and Selection Operator (LASSO) regression with selected lncRNAs. Computational methods like CIBERSORT were employed to quantify immune cell infiltration patterns, and weighted gene co-expression network analysis (WGCNA) was conducted to delineate co-expression modules. Finally, a ceRNA network was constructed to elucidate the regulatory interactions among lncRNAs, miRNAs, and mRNAs.
Results:
We identified 5927 DEGs, among which 47 were differentially expressed long non-coding RNAs (DELs). Seven DELs (DGCR11, FAM215A, HCG9, PART1, FAM106A, NOP14-AS1, PRORY) formed the basis of a classification model with an area under the receiver operating characteristic (ROC) curve of 1. We also examined the infiltration patterns of immune cells in OA tissues and found significant differences compared to healthy controls, indicating a strong immunological component in OA pathogenesis. WGCNA identified a turquoise module with a high correlation to OA traits.
Conclusions:
The study highlights the importance of ceRNA networks in understanding the complex pathogenesis of OA and offers a comprehensive framework for future research into potential diagnostic biomarkers and therapeutic targets.
| eISSN: | 1896-9151 |
| ISSN: | 1734-1922 |
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.