Introduction
An adrenalectomy is a surgical resection of an adrenal gland. Patients are most commonly qualified for adrenalectomy due to a diagnosis of an adrenal tumor, either symptomatic or incidentally detected during imaging studies (so-called incidentaloma). Symptoms of an adrenal tumor may be associated with its size (a mass effect) or an excessive secretion of hormones [1]. Indications for adrenalectomy may therefore be divided into two large groups, namely endocrine and oncologic ones. Endocrine indications include basically all hypersecretion syndromes: Cushing’s syndrome (ACTH-independent or ACTH-dependent, but with contraindications or refractory to transsphenoidal surgery), primary hyperaldosteronism (Conn’s syndrome), hyperandrogenic syndrome of adrenal origin (very rare) and a pheochromocytoma (PHEO; or even a suspicion of one). Oncologic indications appear when an adrenal tumor is of high risk of being malignant, either metastatic or primary. Adrenal glands are common sites of metastases, while primary adrenal malignancies (adrenocortical carcinoma (ACC) and malignant PHEO)) are less common. Oncologic recommendations for adrenalectomy include certain radiologic features of a tumor (high density in unenhanced CT (> 30 Hounsfield units (HU)), slow contrast medium washout (< 50% in the 10th minute) and low lipid content in MR) as well as tumor size greater than 5 cm and/or its fast growth assessed during consecutive imaging studies [2–5].
Preoperative preparations for adrenalectomy depend on the initial diagnosis. Patients diagnosed with hypersecretion syndromes require specific management, which consists of normalization of blood pressure and heart rate using α- and (sometimes) β-blockers (PHEO), control of hypercortisolemia with steroidogenesis inhibitors, e.g. ketoconazole (Cushing’s syndrome), or normalization of blood pressure and hypokalemia using spironolactone and potassium (Conn’s syndrome) [2, 6].
Following the operation, the histopathological examination of the excised specimen ultimately determines the tumor’s character and verifies the preoperative diagnosis. The scope of pathological diagnoses varies widely, including more than a dozen entities, with benign adrenocortical adenoma (ACA) being the most common one [1, 6, 7]. Various pathologic techniques can be used, ranging from classic staining with hematoxylin and eosin (H-E), through different scales designed to assist in the diagnostic process (e.g. the Weiss system), to immunohistochemistry (IHC), which allows one to stain the specimen with antibodies in order to look for a specific protein (antigen). In the case of an adrenal tumor, the most commonly investigated proteins include melan-A (MART-1), inhibin α, chromogranin A (CgA), synaptophysin, neurofilament, protein S-100, calretinin, vimentin, cytokeratins, D2-40, steroidogenic factor-1 (SF-1, Ad4BP), epithelial membrane antigen (EMA) and glycoprotein HMFG-2.
The aim of our study was to compare the initial and final diagnoses of patients undergoing adrenalectomy to determine the level of coherence between them. Based on these data, we estimated the accuracy (sensitivity and positive predictive value (PPV)) for each diagnosis. We also compared the sex and age of patients in every group.
Material and methods
The data of all patients undergoing adrenalectomy, regardless of operation technique, in the 1st Department and Clinic of General, Gastroenterological and Endocrine Surgery, Wroclaw Medical University (WMU), from 2004 to 2018, were investigated. Patients’ demographic data, the results of preoperative laboratory and imaging studies, as well as postoperative histopathological reports, were gathered in a database.
Prior to admittance to the surgical department, patients were diagnosed in an endocrine department for comprehensive evaluation of their hormonal status and establishing or confirming an indication for surgical treatment. The vast majority of patients were referred to our clinic by the Department and Clinic of Endocrinology, Diabetology and Isotope Therapy, WMU. The standard preoperative evaluation included at least one imaging study (computed tomography was preferred) and the following laboratory parameters: complete blood count (CBC), electrolytes (Na+ and K+), serum cortisol profile, free cortisol excretion in 24-hour urine collection, aldosterone and plasma renin activity (PRA) – both in resting and supine position, aldosterone excretion in 24-hour urine collection, metanephrines in 24-hour urine collection. In selected patients, additional tests were performed, including androgen metabolism markers (dehydroepiandrosterone sulfate (DHEA-S), testosterone, androstenedione, sex hormone-binding globulin (SHBG)), dexamethasone test (with small (2 mg) or large (8 mg) dose), adrenocorticotropic hormone (ACTH), catecholamines in plasma and excretion in 24-hour urine collection, vanillylmandelic acid (VMA), chromogranin A (CgA). Based on clinical presentation, imaging studies and laboratory test, patients were qualified for surgical treatment in 8 categories: 1) tumor without hormonal hypersecretion, 2) adrenal cyst, 3) pheochromocytoma or suspicion of one, 4) Cushing’s syndrome, 5) primary hyperaldosteronism (Conn’s syndrome), 6) Conn’s syndrome coexisting with hypercortisolemia, 7) metastatic tumor or suspicion of one and 8) recurrence of previously removed adrenal tumor.
After adrenalectomy, the postoperative specimens were fixed with 10% formalin, transferred to the Department and Division of Pathomorphology, WMU, then processed into paraffin-embedded blocks. Slices from blocks were evaluated using classic H-E staining, as well as IHC techniques, depending on the pathologist’s choice. Definitive pathological diagnoses included 14 positions, namely: (benign) ACA, nodular hyperplasia (NH), ACA/NH (distinction impossible), PHEO, adrenal gland without any detected pathology, myelolipoma, hematoma, adrenal cyst, hemangioma, ganglioneuroma; and (malignant) metastasis, ACC, lymphoma and schwannoma.
When evaluating the PPV and sensitivity for specific preoperative diagnoses, the following assumptions were made: 1) a non-functioning tumor can turn out to be anything on postoperative pathology, apart from an adrenal gland with no pathology or a cyst; 2) a cyst, a pheochromocytoma and a metastasis can only be a cyst, a pheochromocytoma or a metastasis, respectively; 3) Cushing’s or Conn’s syndrome or both combined can be an ACA, NH, ACA/NH or ACC; 4) a recurrent tumor can be any type of malignancy. The assessment of sensitivity was possible only for those preoperative diagnoses that had the same postoperative equivalent, that is: a cyst, PHEO and metastasis (as only then could a false negative (FN)) value be calculated).
Statistical analysis
For statistical comparisons of patients’ sex, the χ2 test was used. The Shapiro-Wilk test was used to check the normality of distribution of the age of patients. For further comparisons of the age of patients between groups, the Kruskal-Wallis test with post-hoc Conover test was used.
Results
There were 214 patients undergoing adrenalectomy from January 2004 to December 2018 in the studied department and clinic. There were 150 (70.1%) females and 64 (29.9%) males, from 21 to 81 years old (mean age: 56.0 ±11.3). The majority of patients underwent a single resection, but 6 of them were operated on twice and one was operated on three times (222 operations in total). The laparoscopy was the preferred surgical approach (141 procedures, 63.5%), followed by laparotomy (55, 24.8%), laparoscopy with conversion to laparotomy (15, 6.8%), retroperitoneoscopic (9, 4.1%) and open extraperitoneal method (2, 0.9%). The left adrenalectomy was slightly more common than the right one (109 (49.1%) vs. 105 (47.3%)); 8 (3.6%) procedures were a simultaneous bilateral adrenalectomy. Therefore 230 postoperative specimens were evaluated in total. Among this group there were no cases of a malignant PHEO or an androgen-producing tumor. Perioperative complications that accompanied some of these procedures were described in the previous paper [8].
The comparison between preoperative and postoperative diagnoses is summarized in Table I. Specimens indicated as non-functioning tumors had a variety of pathological diagnoses, both benign and malignant. The ACA was the most common one (62, 48.8%), followed by NH (14, 11.0%) and – interestingly – myelolipoma (13, 10.2%). Only 8 cases did not match the assumed scope of diagnoses – 3 adrenal glands without any pathology and 5 cysts; therefore the PPV was 93.7%. Three specimens diagnosed as adrenal cysts had their diagnosis confirmed (PPV 100%). However, for 8 cysts recognized in total, only 3 were described as one before an operation (sensitivity 37.5%). Out of 35 cases indicated as PHEO, only 21 were confirmed by pathology (PPV 60.0%). At the same time, for 31 PHEOs diagnosed in total, only 21 had the same preoperative diagnosis (sensitivity 67.7%); the remaining 10 were marked as non-functioning tumors. In the Cushing’s syndrome group, there was only 1 mismatch per 26 (1 adrenal gland without pathology; PPV 96.2%). Similarly in the Conn’s syndrome group, there were 2 mismatches per 20 (1 adrenal gland without pathology and 1 hematoma; PPV 90.0%). One single case of Conn’s syndrome with coexisting hypercortisolemia turned out to be an adrenal gland without pathology. Out of 17 cases of suspected metastases 14 were confirmed (PPV 82.4%), yet 3 actual metastases were initially classified simply as non-functioning tumors (sensitivity 82.4%). The origin of metastases was: lung cancer (7 cases, 41.2%), renal cancer (4, 23.5%), adenocarcinoma (site unknown; 2, 11.8%) digestive tract neoplasm (suspicion; 2, 11.8%), skin melanoma (1, 5.9%) and neoplasm of unknown origin (1, 5.9%). One single case of a recurrence of a tumor in a lodge of a previously removed adrenal gland was an ACC; the pathological report from the first procedure was “borderline adenoma” and a close clinical follow-up was indicated. Out of 3 recognized ACCs, 2 were initially diagnosed as non-functioning tumors.
Table I
Comparison of preoperative and postoperative diagnoses
[i] TU – non-functioning tumor, PHEO – pheochromocytoma, C + C – Conn’s and Cushing’s syndrome, META – metastasis, REC – recurrence of a removed tumor, AG w/o pathol. – adrenal gland without any detected pathology, TP – true positive, FP – false positive, FN – false negative, PPV – positive predictive value; diagnostic mismatches are marked by filled (grey) boxes.
A high PPV signifies a high chance of confirmation of preoperative diagnosis. Most of them were characterized by a high PPV: adrenal cyst and recurrent tumor – both 100%, Cushing’s syndrome – 96.2%, non-functioning tumor – 93.7%, Conn’s syndrome – 90.0%, suspected metastases – 82.4%. Only pheochromocytoma had considerably lower PPV (only 60.0%), indicating that the remainder (40.0%) were over-diagnosed.
The lower the sensitivity, the greater was the fraction of patients with said condition that was not properly diagnosed (under-diagnosed). Suspected metastases had the highest value (82.4%), followed by pheochromocytoma (67.7%), and adrenal cyst (37.5%).
Regarding the initial (preoperative) diagnosis, there was a statistically significant difference in terms of patients’ sex (p = 0.001) – Table II. Further analysis showed that the diagnosis of Cushing’s syndrome was more common in females (p = 0.009) and, in turn, the diagnosis of metastases to adrenal glands was more common in males (p = 0.001). There was also a statistically significant difference in terms of patients’ age (p = 0.008). Patients diagnosed with Conn’s syndrome were significantly younger than those diagnosed with PHEO (p = 0.005), non-functioning tumor (p = 0.002) and recurrent tumor (p = 0.043). Patients diagnosed with a recurrent tumor were also older than those diagnosed with an adrenal cyst (p = 0.045) and simultaneous Cushing’s and Conn’s syndrome (p = 0.028). Patients diagnosed with non-functioning tumor were older than those with Cushing’s syndrome (p = 0.044).
Table II
Demographic characteristics of studied groups
Regarding the final (postoperative) diagnosis, there was a statistically significant difference in terms of patients’ sex (p = 0.016). Metastases to adrenal glands were more common in males (p = 0.001). There was no statistically significant difference in terms of patients’ age (p = 0.112).
Discussion
The majority of patients in our study were qualified for an adrenalectomy due to a non-functioning adrenal tumor (127/230; 55.2%). Specific indications for surgical resection included: sufficiently large tumor size (≥ 4 cm, associated with an increased risk of malignancy), its fast growth in consecutive imaging studies, high density in CT (not typical for a benign adenoma) or other suspicious CT features, such as calcifications or necrosis – Figures 1 A and B [5, 9]. Most of the tumor in this group (116/127; 91.3%) turned out to be benign in postoperative histopathological evaluation. Polish guidelines from 2002 recommend surgical removal of an adrenal tumor measuring ≥ 4 cm and such an approach was taken in most of the cases described in this paper. Yet an American algorithm (National Institute of Health, 2003) allows observation of such tumors, recommending adrenalectomy only for tumors ≥ 6 cm, unless other radiological features of malignancy are present. This relies on data indicating that the estimated risk of malignancy is about 2% in an adrenal tumor < 4 cm, but rises up to 25% in lesions ≥ 6 cm [10]. Similarly, the updated Polish guidelines from 2016 recommend 5 cm to be a more suitable cut-off value, but only some portion of the studied patient group was operated on after 2016 [2]. We also detected that patients in the non-functioning tumor group were older than those in Cushing’s or Conn’s syndrome groups (p = 0.044 and p = 0.002, respectively).
Figure 1
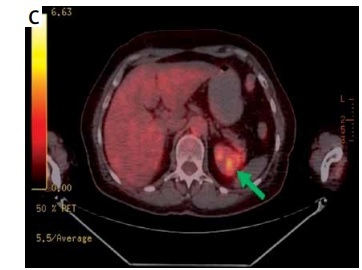
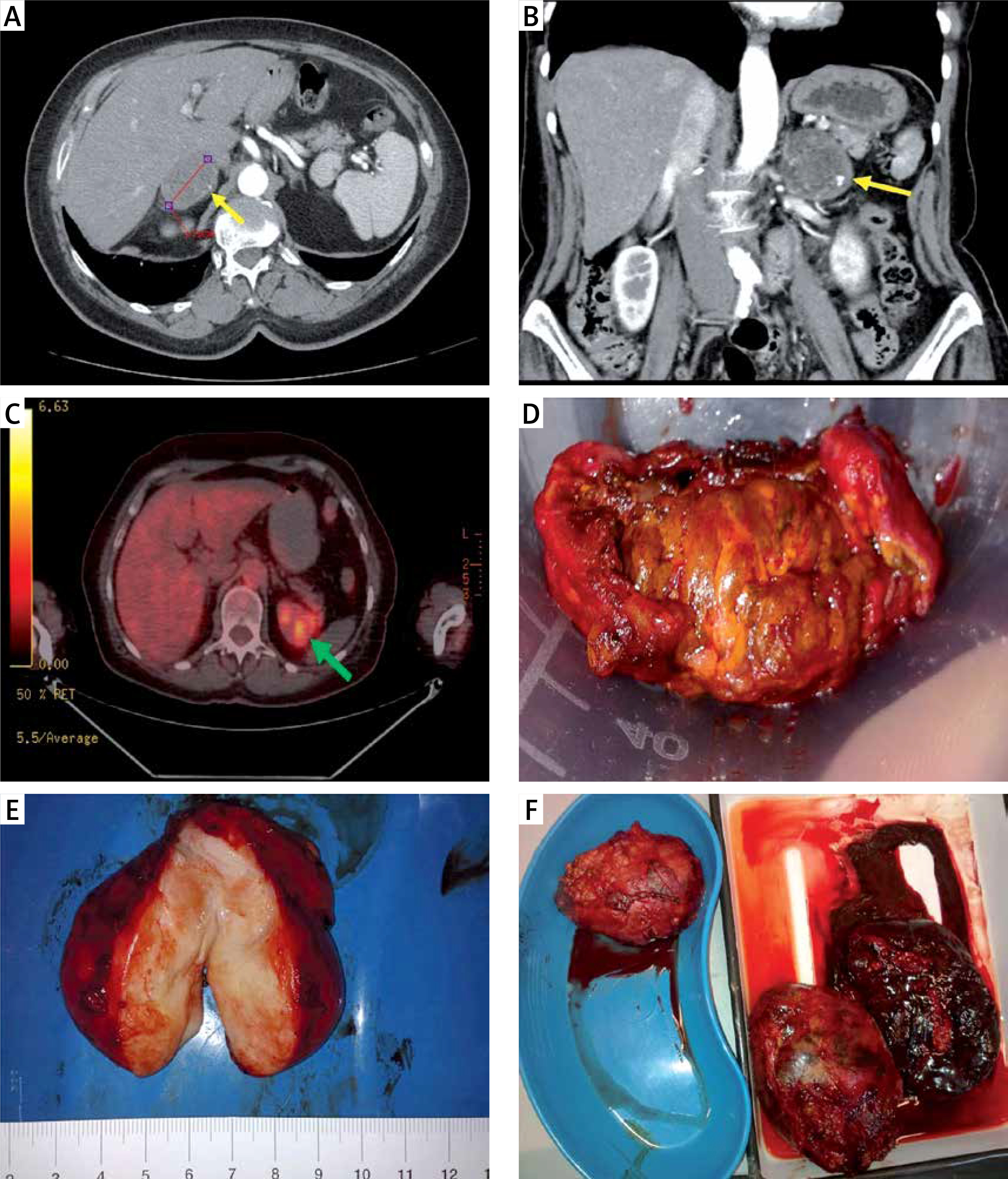
Preoperative imaging studies and postoperative specimens of adrenal tumors. A – Computed tompography (CT), transverse scan: right adrenal gland tumor, 47 mm in greatest dimension, 72-year-old woman; postoperative histopathologic report: hematoma of a medullar part. B – CT, coronal scan: left adrenal gland tumor, 50 mm in greatest dimension, heterogenicity and calcifications seen within the tumor; 69-year-old woman; postoperative histopathologic report: adrenocortical adenoma 5 × 4 × 2,5 cm. C – PET-CT, transverse scan: left adrenal gland tumor, 85 mm in greatest dimension, suspicion of metastasis (73-year-old man who underwent right nephrectomy 5 years before due to clear-cell renal cell carcinoma (CCRCC)); postoperative histopathologic report: CCRCC metastasis to adrenal gland. D – Adrenocortical adenoma (typical golden color), around 4 cm, 52-year-old woman with Cushing’s syndrome. E – Ganglioneuroma, around 8 cm, 43-year-old woman with non-functioning tumor. F – Metastases of lung cancer to both adrenal glands, around 10 and 11 cm (size comparable to a spleen, shown in a lower right corner), 67-year-old man
The second main indication for adrenalectomy was hypersecretion of an adrenal cortex (47/230; 20.4%). Patients qualified due to Cushing’s syndrome were significantly more often females (p = 0.009). Patients in the Conn’s syndrome group were significantly younger than those in PHEO (p = 0.005), non-functioning tumor (p = 0.002) and recurrent tumor groups (p = 0.043). The important fact in a preoperative diagnosis of Conn’s syndrome is the discontinuation of specific drugs (including ACE inhibitors, angiotensin receptors blockers, progestogens, diuretics, spironolactone and eplerenone) for up to several weeks prior to any hormonal workup, since these substances may alter the aldosterone-to-renin ratio (ARR) [2, 5, 10].
The final determination as to the character of an excised adrenal mass is established by a histopathological examination [6, 11]. The scope of possible diagnoses includes at least a dozen options.
Adrenocortical adenoma (ACA) is the most frequent diagnosis (70–94%). Typically ACAs are small (less than 50 g in weight), well-separable from surrounding tissues and golden/yellow in color (Figure 1 D). Most ACAs are hormonally inactive. Among hormonally active ACAs, aldosterone-producing ACAs (APAs) are more common than cortisol-producing ACAs (CPAs), while androgen-producing ACAs are very rare [1, 4, 7, 12]. This is in accordance with our results: there were 72 (69.2%) hormonally inactive ACAs, 16 (15.4%) APAs, 16 (15.4%) CPAs and no androgen-producing ACAs.
Another form of hormonally active adrenocortical growth is nodular hyperplasia (NH). In our observation there were 20 (71.4%) non-functioning NHs, 6 (21.4%) causing Cushing’s syndrome and 2 (7.1%) causing Conn’s syndrome [13]. In 3 cases discrimination between ACA and NH was impossible due to fragmentation of a specimen.
In a group referred to as non-functioning tumors, the third most commonly recognized pathology was myelolipoma (13 cases, 10.2%). This was similar to the results of Al Harthi et al., who estimated the detection of this pathology to be 7% of adrenal incidentalomas. To date it remains unknown how this kind of tumor actually develops. It is usually a small, asymptomatic and non-functional lesion, a benign neoplasm containing mature adipose tissue and hemopoietic elements (but not producing actual blood cells) [14].
PHEO is usually a benign, sporadic, catecholamine-producing adrenal tumor. However, in about 9–23% of cases (and even more in children) it develops in extraadrenal tissues close to sympathetic ganglia (so-called paraganglioma). Also up to 25% of patients may present with the hereditary form of PHEO (e.g. in the course of multiple endocrine neoplasia type 2 (MEN-2) or von Hippel-Lindau (VHL) syndrome), with a tendency for extraadrenal and multifocal tumors. Additionally, in up to 26–35% of cases it is malignant, and at the time of diagnosis, about 10% of patients with PHEO present with a metastatic (disseminated) disease [15]. This is in opposition to our findings, as we did not observe a single case of malignant PHEO. Perhaps such patients were referred to other departments for surgical treatment. According to Myśliwiec et al., biochemical diagnosis of PHEO is associated with a significant percentage of false positive (FP) results, reaching 33%. We can fully substantiate that statement, as in our observation PHEO had the lowest PPV (60.0%), making the remaining 40.0% of cases FP (over-diagnosed). Such a high amount of FP diagnoses may result from various possible distractions in the diagnostic process. Diverse substances, including meals (bananas, chocolate, cocoa, citrus fruits, vanillin), drinks (coffee, tea), drugs (e.g. α- and β-blockers, sympathicomimetics, calcium channels antagonists, aminophylline, disulfiram, L-DOPA, methyldopa, insulin, tetracycline, erythromycin) and narcotics (cocaine, amphetamine, (pseudo)ephedrine) may artificially elevate levels of urinary metanephrines and should definitely be discontinued before performing such diagnostics. The next reason for overdiagnosis may be a routine evaluation of urinary 24-hour metanephrines only and not plasma free metanephrines in a suspicion of PHEO. Testing for plasma metanephrine levels is slightly more sensitive and specific, yet it is also more expensive and less available; therefore the urinary metanephrines remain the most commonly performed test. Moreover, the test for plasma metanephrines became available just a few years ago and our research dates back before this time. The diagnosis of PHEO should also be supported by imaging studies – in a CT PHEO exhibits a non-adenoma pattern (higher density: > 10 HU, usually > 30 HU, compared to < 10 HU for adenoma) [2, 10, 16]. One last issue here is the human factor, that is the fear of omission (under-diagnosis) of PHEO. Since it may be associated with serious complications, physicians are anxious about overlooking a single case of PHEO and may be prone to rather exaggeratedly qualify a patient for an operation than to risk letting go a dubious case.
Adrenal cysts are diagnosed in about 5–8% of incidentalomas, usually unilaterally (> 80%) and more commonly in females – as was the case in our observations (5 females and 3 males). Several types of cysts can be distinguished, including endothelial (congenital or retentive, hemangiomas and lymphangiomas), epithelial (associated with a parasitic infection) and pseudocysts (usually resulting from a hemorrhage or necrosis). Some fraction of lesions classified as hemorrhagic pseudocysts may originate from an angioma, which structure has been destroyed by an internal hemorrhage. Adrenal cysts are associated with about 7% risk of malignancy [17, 18]. In our study a diagnosis of an adrenal cyst had a perfect PPV (100%), yet a relatively poor sensitivity (37.5%), indicating that most of these lesions are initially recognized as solid non-functioning tumors.
Ganglioneuroma is a rare, benign, and typically asymptomatic tumor of the autonomic nerve fibers (consisting of ganglion cells, Schwann cells and fibrous tissue). Apart from adrenal glands, it may be found within the retroperitoneal space, posterior mediastinum, head and neck. Macroscopically it is usually a firm, solid, white tumor (Figure 1 E). Only symptomatic cases require surgical treatment. We had 2 ganglioneuromas in our observation, both initially diagnosed as non-functioning adrenal masses.
Adrenal glands are common sites of metastases from various malignancies of other locations. In about 0.2% of cases an adrenal incidentaloma turns out to be the only manifestation of a previously undiagnosed cancer. If an extra-adrenal malignancy is present, the risk of a co-existing adrenal tumor being malignant increases from 0.1% to 50–75% [7]. According to the literature, the most common sources of metastases to adrenal glands are: lung, renal, breast, gastrointestinal tract (gastric, colorectal) and liver cancers (hepatocellular carcinoma – HCC) as well as skin melanoma [19]. We observed similar proportions, as the source of most metastases was lung cancer (7; 41.2%), followed by renal cancer (4; 23.5%; Figure 1 C), adenocarcinoma (site unknown; 2; 11.8%) digestive tract neoplasm (2; 11.8%), skin melanoma (1; 5.9%) and a neoplasm of unknown origin (1; 5.9%). In our observation the metastasis group had a high PPV as well as sensitivity (both 82.4%). We also detected that metastases were diagnosed predominantly in males, both pre- and postoperatively (both p = 0.001). Taking into account that the most common sources of metastases are lung and renal cancer, which are more prevalent in males, this seems justified. Metastatic adrenal tumors may reach especially large dimensions, such as those of lung cancer presented in Figure 1 F, measuring 10 and 11 cm (comparable to the removed spleen).
ACC is a malignant neoplasm originating from an adrenal cortex. It is found mostly in older females, as we observed (2 females vs. 1 male; mean age 68.0 ±12.4). Despite its rarity (incidence 0.5–2/million/year), ACC is an aggressive neoplasm, lacking early symptoms and therefore often diagnosed at an advanced stage and associated with a poor prognosis. More than half of patients (43–69%, mean 54%) present with stage III/IV and almost half of them (38–40%) have metastases; therefore the overall 5-year survival ranges between only 16 and 44% (mean: 30%) [7]. In our study 2 ACCs were found in the non-functioning tumor group (constituting 1.6% of this group) and 1 was initially diagnosed as a recurrent adrenal tumor (100% of this group). The regrowth of a previously resected adrenal tumor should always raise a suspicion of a malign neoplasm.
To sum up, the advantage of our study is a presentable group of examined patients and specimens (214 and 230, respectively), since adrenalectomy is not routinely performed in local primary care surgical centers and such material is not easy to obtain. On the other hand, the limitation of our study is both the retrospective design and a significant time period of observation (2004–2018), during which much has changed in the area of qualifications as well as surgical approach to adrenalectomy.
In conclusion, the majority of diagnoses of adrenal tumors are characterized by both high PPV and sensitivity. Yet PHEO is characterized by both much lower PPV and sensitivity (60.0% and 67.7%, respectively), indicating that many cases are either over- or under-diagnosed, and closer preoperative diagnostics are necessary to improve these rates. Meticulous preparation of a patient for hormonal tests, including discontinuation of certain medications, is essential for obtaining accurate results. The diagnosis of an adrenal cyst is always confirmed postoperatively (PPV 100%), but some masses initially recognized as solid tumors also turn out to be cysts (sensitivity: 37.5%). Patients qualified for an adrenalectomy due to Cushing’s syndrome are significantly more often females (p = 0.009), while those with metastases (diagnosed both pre- and postoperatively) are more often males (both p = 0.001). Patients qualified due to non-functioning tumors are older than those with Cushing’s or Conn’s syndrome (p = 0.044 and p = 0.002, respectively). Adrenocortical carcinoma may initially be diagnosed as a non-functioning tumor (1.6% of such cases) or a recurrence of a previously resected tumor, which should always raise a suspicion of a malignant neoplasm.