Introduction
Lipopolysaccharide (LPS) from the outer membrane of bacteria is the primary trigger of the systemic inflammatory response in sepsis [1]. LPS could mediate several inflammatory responses during sepsis and septic cardiomyopathy [2, 3] and may participate in the contractile dysfunction observed in chronic heart failure [4–6]. Prolonged exposure (over 24 h) of adult rat ventricular myocytes to a mixture of LPS and inflammatory cytokines inhibited cell contractility [7]. Interestingly, LPS at a low concentration increased sarcomere shortening, shortening velocity, and relaxation velocity in cardiac myocytes [7]. Therefore, the divergent effects of LPS have also been characterised in ventricular myocytes isolated from adult rats.
Toll-like receptor 4 (TLR4) is required for cardiac myocyte impairment during endotoxaemia [8], and TLR4 is known as an LPS binding site [9]. Additionally, the Janus-activated kinase 2 (JAK2) and signal transducer and activator of transcription3 (STAT3) pathway (JAK2/STAT3 pathway) are coupled to signalling through cytokine receptors including TLR4 [10]. Interestingly, an agent was discovered that improves left ventricular performance via activation of the JAK2/STAT3 pathway in rats [11]. LPS-induced cardiac contraction has also been observed in dogs [12] and calves [13]. The LPS-induced alterations in cardiac function in mice are indicated to be due to altered loading conditions but not to reduced contractility, which may instead be slightly increased [14].
The effects of LPS on cardiac performance were shown to have two patterns in vitro, resulting in enhanced performance at low concentrations and inhibited performance at high concentrations [7]. Therefore, understanding the variations in LPS-induced cardiac injury in detail is of interest.
In the present study, we investigated the direct effects of LPS on the cardiac contractility of isolated ventricular myocytes using a Langendorff apparatus. Changes in the relevant signalling pathways were also characterised in H9c2 cells exposed to LPS.
Material and methods
Material
Lipopolysaccharide from Escherichia coli 0111: B4 was purchased from Sigma-Aldrich (St Louis, MO, USA). The fluorescent probe fura-2 was from Molecular Probes (Eugene, OR, USA). Antibodies to cardiac contractile protein myosin light chain 2 (MLC2) and phospho-myosin light chain 2 (pMLC2) (p-Ser19) were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Experimental animals
Male Sprague-Dawley (SD) rats weighing 250 to 280 g were obtained from the National Laboratory Animal Centre (Taipei, Taiwan). They were housed individually in plastic cages under standard laboratory conditions. The animals used in all the experiments were maintained under anaesthesia with sodium pentobarbital (35 mg/kg, i.p.) to minimise suffering. The experimental protocols were approved by the Institutional Animal Ethics Committee (103101525) of Chi-Mei Medical Centre. All experiments conformed to the Guide for the Care and Use of Laboratory Animals as well as the guidelines of the Animal Welfare Act.
Langendorff apparatus for isolated heart determination
The experiments were performed according to our previous method [15]. Rats in each group were sacrificed under anaesthesia with 3% isoflurane, and their hearts were excised rapidly and rinsed by immersion in ice-cold Krebs-Henseleit buffer (KHB). The isolated ventricular myocytes mounted on the Langendorff apparatus were continuously perfused with warm (37°C) and oxygenated (95% O2, 5% CO2) KHB at a constant pressure of 70 mm Hg. The organ chamber temperature was maintained at 37°C during the experiment. A water-filled latex balloon was inserted through an incision in the left atrium into the left ventricle via the mitral valve and adjusted to a left-ventricular end-diastolic pressure (LVEDP) of 5–7 mm Hg during initial equilibrium. The distal end of the catheter was connected to the data acquisition system via a pressure transducer for continuous recording. The left-ventricular developed pressure (LVDP) was obtained from the difference between left-ventricular systolic pressure (LVSP) and LVEDP. Heart rates were monitored simultaneously. In each experiment, after a 30-min stabilisation with perfusion, the tested agent including LPS at the desired concentration was added into the KHB for further perfusion.
Cell culture
H9c2 cells (BCRC No. 60096), a cardiac myoblast cell line, were cultured following our previous method [16]. Briefly, H9c2 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, pH 7.2; GIBCO-BRL Life Technologies, Gaithersburg, MD, USA) supplemented with 10% foetal bovine serum. Cells were passaged every 3–4 days and subcultured when they reached 70–80% confluency. For H9c2 differentiation, cells were switched to differentiation medium (1% FBS-containing medium with 1 μM trans-retinoic acid, RA) for seven days as described previously [17]. After plating, the medium was replaced on the second day. The following day, the cells were incubated with LPS and/or inhibitors, as subsequently described.
Drug administration
In each experiment with the Langendorff apparatus, after 30 min of stabilisation with perfusion, the tested agent including LPS at the desired concentration was added to the solution for further perfusion. Samples were pretreated with inhibitors for 10 min before perfusion with LPS. Cultured cells were pretreated with the specific inhibitors at the indicated doses for the desired time. Then, H9c2 cells were incubated with LPS at various doses for a 3-h or 24-h period, as described in each assay.
Measurement of changes in intracellular calcium levels
We measured intracellular calcium levels using fura-2, a fluorescent probe, as described previously [18]. Changes in fluorescence were recorded using a fluorescence spectrofluorometer (F-2000; Hitachi, Tokyo, Japan). The intracellular calcium [Ca2+] i values were estimated according to our previous report [18]. Background autofluorescence obtained from untreated cells was subtracted from all measurements. Additionally, inhibitor effectiveness was assessed after a 30-min pretreatment.
Flow cytometry analysis
H9c2 apoptotic cell measurements were performed by flow cytometry with Annexin V-propidium iodide staining, as described previously (30), using a fluorescein isothiocyanate (FITC) apoptosis detection kit (BD Biosciences, San Diego, CA, USA). The Annexin reagent utilises Annexin V to detect phosphatidylserine on apoptotic cells and a dead cell marker as an indicator of membrane stability. H9c2 cells were seeded in a dish (10 cm) at a density of 1 × 106 cells/well, and the experimental procedure was performed according to the manufacturer’s protocol. Staining was analysed by fluorescence-activated cell sorting on a flow cytometer (NOVO Cyte 3000; ACEA Biosciences, San Diego, CA, USA). Spectral compensation and data quantification were performed using the NovoExpress software (2017 Version; ACEA Biosciences Inc., San Diego, CA, USA), which provides a variety of plots and gates for flow cytometry data analysis.
Determination of LDH release
LDH is a marker for remaining cells. To explore LPS-induced LDH release, H9c2 cell supernatants were collected for LDH determination in serum-free medium. LDH levels were measured in cell culture medium (20 μl) at different time points using a commercial kit (Biovision, Milpitas, CA, USA), as described previously [19]. The results were expressed as the percentage of total LDH activity (total LDH activity = LDH activity in the cell-free medium + LDH activity in the cell medium). Released LDH was calculated according to the following equation: LDH released (%) = (LDH activity in the serum-free medium/total LDH activity) × 100.
Real-time reverse transcription-polymerase chain reaction
Total RNA was isolated from H9c2 cardiomyocytes using TRIzol followed by chloroform extraction. The extracted messenger RNA (2 μg per sample) was reverse transcribed into cDNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland), according to the manufacturer’s instructions.
Real-time quantitative polymerase chain reaction (RT-PCR, qPCR) was performed using SYBR Green (Roche) and mouse-specific primers on a LightCycler 480 system. The signal intensity was normalised to GAPDH. The primers used in this study are shown below.
TLR4 F: 5′-CATGGCATTGTTCCTTTCCT-3′;
TLR4 R: 5′-CATGGAGCCTAATTCCCTGA-3′;
ANP F: 5′-CACAGATCTGATGGATTTCAAGA-3′;
ANP R: 5′-CCTCATCTTCTACCGGCATC-3′;
BNP F: 5′-GTCAGTCGCTTGGGCTGT-3′;
BNP R: 5′-CCAGAGCTGGGGAAAGAAG-3′;
β-MHC F: 5′-CATCCCCAATGAGACGAAGT-3′;
β-MHC R: 5′-GGGAAGCCCTTCCTACAGAT-3′;
CaSR F: 5′-CCTGCTGGGACTTTTCTACATC-3′;
CaSR R: 5′-TGTACTGGTTCTTATTGCTGAGGA-3′;
IL6 F: 5′- GATGAGTACAAAAGTCCTGATCCA-3′;
IL6 R: 5′- CTGCAGCCACTGGTTCTGT -3′;
TNF-α F: 5′-CAGCCTCTTCTCCTTCCTGAT-3′;
TNF-α R: 5′-GCCAGAGGGCTGATTAGAGA-3′;
β-actin F: 5′-CTAAGGCCAACCGTGAAAAG-3′;
β-actin R: 5′-GCCTGGATGGCTACGTACA-3′
Western blotting analysis
Total proteins (30 μg) were separated by SDS/polyacrylamide gel electrophoresis (10% acrylamide gel) using the Bio-Rad Mini-Protein II System. Proteins were transferred to expanded polyvinylidene difluoride membranes (Pierce, Rockford, IL, USA). Following blocking, the membrane was probed with the primary antibodies. The blots were incubated with goat polyclonal antibody (1 : 1000) to bind actin, which served as the internal control. After removal of the primary antibody, the blots were incubated for 2 h at room temperature with the appropriate peroxidase-conjugated secondary antibody and then developed by autoradiography using an ECL-Western blotting system (Amersham International, Buckinghamshire, UK). The immunoblots for MLC2 (19 kDa) and phospho-MLC-2 (19 kDa) were quantified with a laser densitometer.
Statistical analysis
Results are presented as the means ± SEM of the sample number (n) for each group. Analysis of variance (ANOVA) was used to evaluate the significance of differences between multiple groups. Once significance was established between the groups, Tukey’s post hoc analysis was used. Values of p < 0.05 were considered significant.
Results
Effects of LPS on the contractility of isolated ventricular myocytes mounted on a Langendorff apparatus
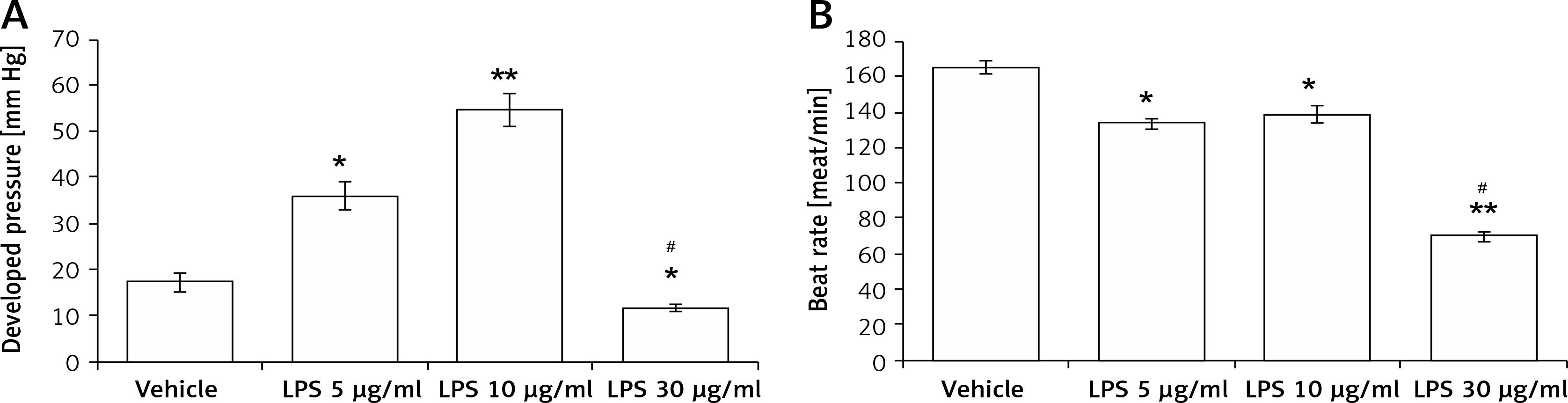
The contractile functions in isolated hearts were increased after perfusion with LPS at 5 μg/ml (2.3 ±0.3-fold) and 10 μg/ml (3.8 ±0.6-fold) compared with that in vehicle-treated controls. However, LPS at 30 μg/ml was shown to decrease the cardiac contractility (0.7 ±0.1-fold, Figure 1 A). Similarly, LPS at 5 μg/ml and 10 μg/ml enhanced the heart rate, while LPS at 30 μg/ml reduced the heart rate (Figure 1 B). The results indicated that LPS promotes cardiac contraction and beating at a low dose but attenuates heart functions at a higher dose.
Figure 1
Direct effects of LPS on cardiac contractility in isolated rat hearts using a Langendorff apparatus. Changes in left ventricular function after treatment with LPS at different concentrations in a Langendorff-perfused heart. A – Quantification of contractile function. B – Beat rate
*P < 0.05 and **p < 0.01 vs. the vehicle-treated control group. #P < 0.05 vs. the 5 μg/kg LPS-treated group, n = 6.
Effects of LPS on the TLR4 signalling pathway in H9c2 cells
In an attempt to elucidate the signalling pathway of TLR4, H9c2 cells were directly incubated with LPS. Clear signals could be obtained only after 30 min of LPS exposure. Then, we compared the LPS-treated cells with the vehicle-treated controls. The representative responses of each signal determined from the Western blots are shown in Figure 2 A. Quantification of the changes in each signal is presented in Figures 2 B–D.
Figure 2
Direct effects of LPS on the TLR4 signalling pathway in H9c2 cells. A – Representative pictures of Western blots, with each signal indicated. Quantified protein levels were calculated for TLR4 (B), JAK2 (C), and STAT3 (D). E – Changes in the cardiac contractile protein myosin light chain (MLC)-2 were compared. F – LPS (5 μg/ml) increased cellular calcium accumulation, which was suppressed by inhibitors including GIT-27, AG490, and Stattic at the indicated concentrations
*P < 0.05 and **p < 0.01 vs. the vehicle-treated control (first column). #P < 0.05 vs. another LPS-treated group (second column), n = 4.
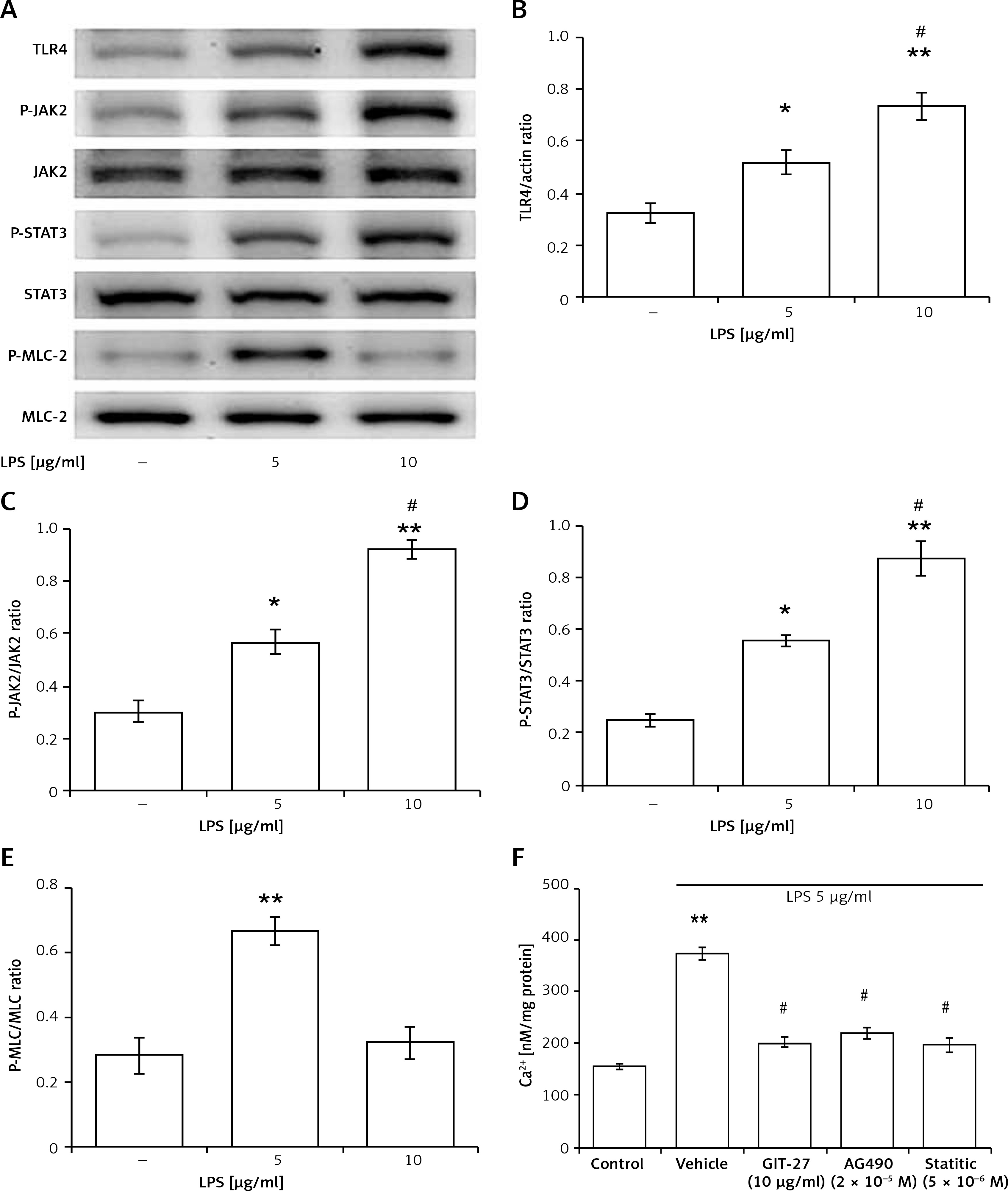
In line with the classically described signalling pathway, phosphorylated JAK2 and phosphorylated STAT3 were increased by treatment with 5 to 10 μg/ml LPS in a dose-dependent manner (Figure 2 A). The quantified data also showed that activated JAK2 and activated STAT3 were increased by LPS treatment in the same manner (Figures 2 C, D). Interestingly, LPS promoted the expression of TLR4 (Figure 2 B) in a manner similar to the changes in mRNA levels. Therefore, LPS may activate TLR4 to induce JAK2 and STAT3 signalling in cardiac myocytes.
However, LPS activated the established contractile protein myosin light chain-2 only at 5 μg/ml. As shown in Figures 2 A and E, levels of phosphorylated myosin light chain-2 were not increased by 10 μg/ml of LPS. This finding indicates that LPS could induce cardiac contraction in H9c2 cells only at 5 μg/ml.
Additionally, we also evaluated the changes in cellular calcium using a fluorescent dye. In H9c2 cells, intracellular calcium levels were dose-dependently increased by 5 to 10 μg/ml LPS. We then investigated the effects of inhibitors specific for each signal on intracellular calcium levels. The calcium accumulation elicited by 5 μg/ml LPS was significantly reduced by each inhibitor. Interestingly, the attenuated levels after treatment with each inhibitor were similar, as shown in Figure 2 F.
LPS exerts time- and dose-dependent effects on cellular function in H9c2 cells
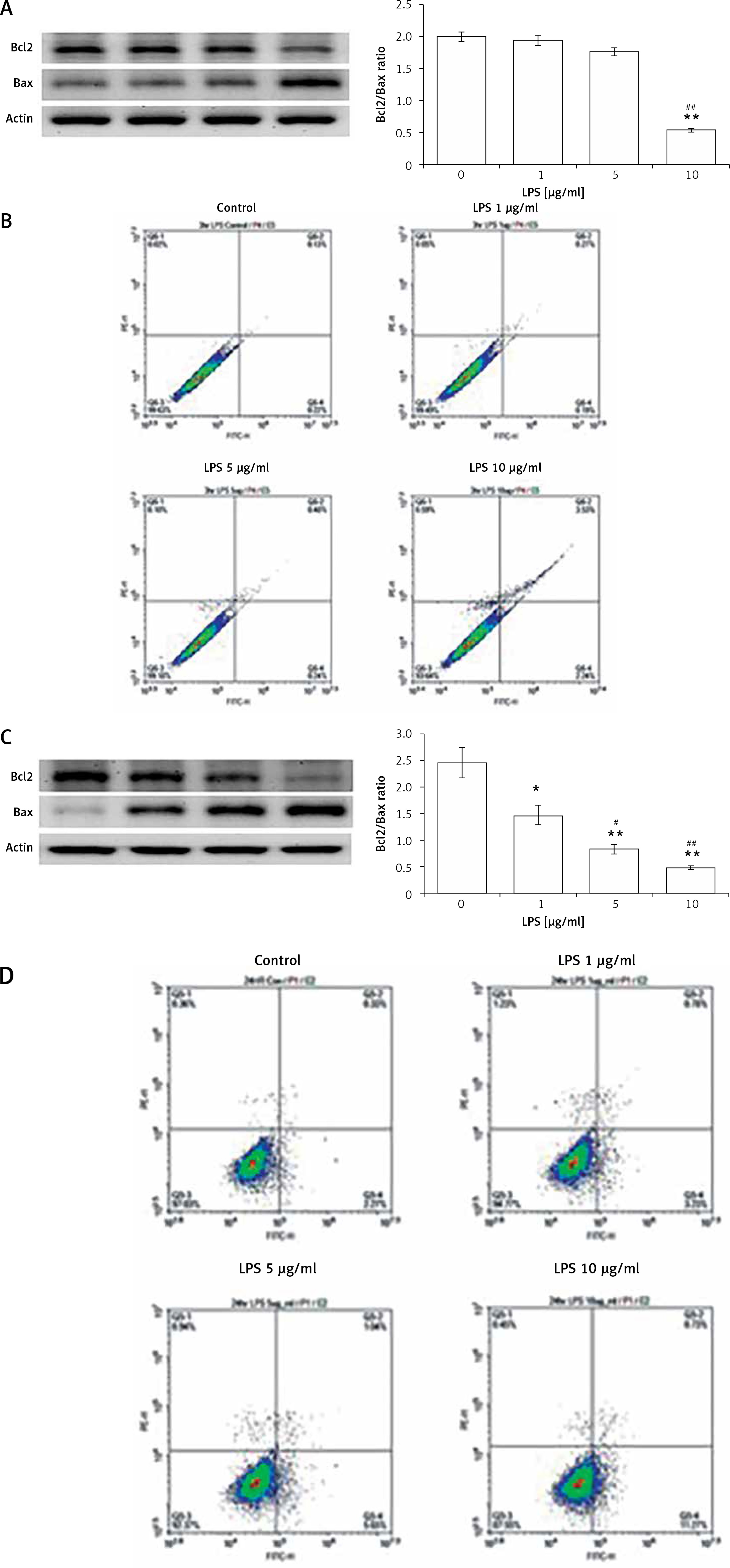
LPS is known to induce cardiac hypertrophy and apoptosis in the heart. We treated H9c2 cells with LPS at various concentrations for two different exposure times. First, H9c2 cells were incubated with 1, 5, and 10 μg/ml LPS for 3 h. Hypertrophic signals were dose-dependently increased by 1 to 5 μg/ml LPS (Table I). However, these signals were all attenuated by 10 μg/ml LPS. Additionally, apoptotic signals, as detected either by Western blot (Figure 3 A) or flow cytometry (Figure 3 B), were induced only by 10 μg/ml LPS. Thus, while a short exposure to a low concentration of LPS may promote hypertrophic signals in the heart, apoptotic signals were observed only when H9c2 cells were exposed to LPS for 24 h. As shown in Table I, hypertrophic signals were reduced by 24-h LPS exposure in a dose-dependent manner. Consequently, apoptotic signals were induced in both Western blot (Figure 3 C) and flow cytometry experiments (Figure 3 D) by 24-h LPS exposure at the same dose.
Table I
Effects of LPS on hypertrophic biomarkers in H9c2 cells after a short (3 h) or longer (24 h) treatment time
| Variable | Control | LPS (1 μg/ml) | LPS (5 μg/ml) | LPS (10 μg/ml) |
|---|---|---|---|---|
| Incubation for 3 h: | ||||
| Relative mRNA of ANP/actin | 1.00 ±0.00 | 1.16 ±0.02* | 1.55 ±0.06**# | 0.73 ±0.06# |
| Relative mRNA of BNP/actin | 1.00 ±0.00 | 1.14 ±0.04* | 1.46 ±0.02**# | 0.82 ±0.02# |
| Relative mRNA of β-MHC/actin | 1.00 ±0.00 | 1.19 ±0.02* | 1.56 ±0.06**# | 0.75 ±0.05# |
| Incubation for 24 h: | ||||
| Relative mRNA of ANP/actin | 1.00 ±0.00 | 0.81 ±0.03* | 0.62 ±0.02**# | 0.39 ±0.03**# |
| Relative mRNA of BNP/actin | 1.00 ±0.00 | 0.85 ±0.02* | 0.59 ±0.03**# | 0.35 ±0.03**# |
| Relative mRNA of β-MHC/actin | 1.00 ±0.00 | 0.84 ±0.03* | 0.61 ±0.02**# | 0.37 ±0.03**# |
Figure 3
Apoptosis induced by LPS in H9c2 cells. Western blot detection of apoptotic biomarkers that were promoted by LPS treatment at the indicated concentrations after a short (3 h) incubation (A) or longer (24 h) treatment (C). The upper panel shows the representative responses, and quantification of the levels is indicated in the last column. Additionally, apoptosis in H9c2 cells was characterised by flow cytometry after a short (3 h) incubation (B) or longer (24 h) treatment (D) with LPS at the indicated dose. The side-scatter intensity (vertical axis) is plotted against the fluorescence intensity in the APC channel (horizontal axis)
*P < 0.05 and **p < 0.01 vs. the vehicle-treated control (first column). #P < 0.05 and ##P < 0.01 vs. the low-dose LPS-treated group (second column), n = 4.
The role of proinflammatory cytokines in the toxic effect of LPS on H9c2 cells
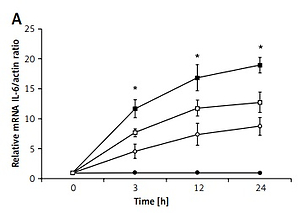
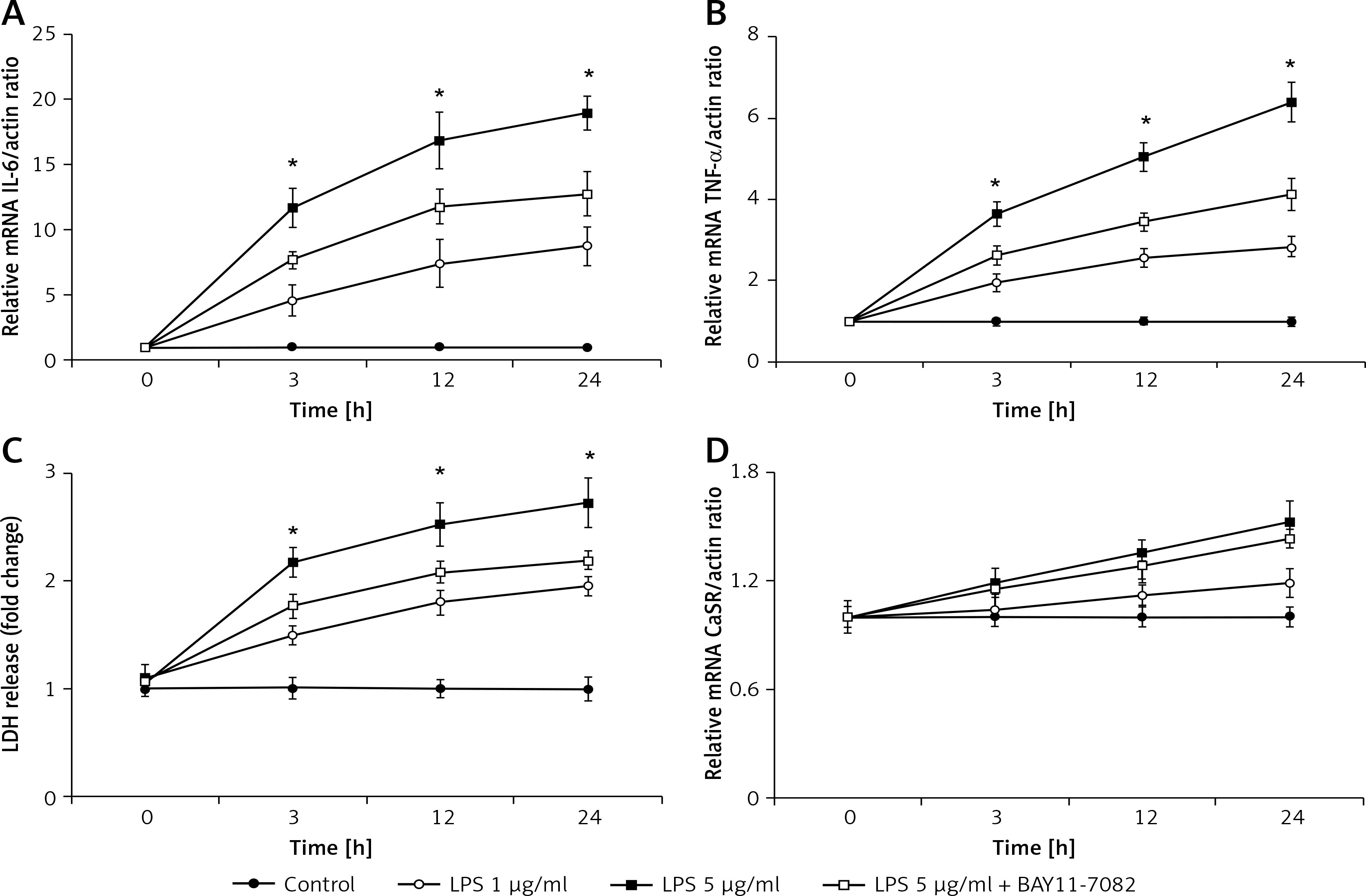
The role of proinflammatory cytokines in apoptosis was investigated using H9c2 cells treated with 24-h exposure to LPS. The expression of interleukin (IL)-6 (Figure 4 A), tumor necrosis factor α (TNF-α) (Figure 4 B), and calcium-sensing receptor (CaSR) (Figure 4 D), as well as LDH release (Figure 4 C), were upregulated by LPS at both concentrations in a dose- and time-dependent manner. BAY11-7082, a potential inflammasome inhibitor, is an irreversible inhibitor of TNF-α-induced IκBα phosphorylation. In the presence of BAY11-7082, the time-dependent changes in IL-6 and TNF-α expression and LDH levels due to LPS at a higher concentration (5 μg/ml) were markedly decreased. However, CaSR expression was not markedly modified by BAY11-7082 in the same manner.
Figure 4
The role of proinflammatory cytokines in the toxic effects of LPS on H9c2 cells. Cells were treated with LPS (1 μg/ml or 5 μg/ml) or vehicle for 3, 12, and 24 h; cells were treated with or without BAY11-7082 (10 μmol/l) 30 mins before LPS (5 μg/ml) treatment. A – IL-6 mRNA expression, B – TNF-α mRNA expression, C – lactate dehydrogenase (LDH) release, D – CaSR mRNA expression
*P < 0.05 was compared between the 5 μg/ml LPS-treated group and the LPS (5 μg/ml), BAY11-7082-treated group, n = 6.
The role of calcium-sensing receptor in the toxic effect of LPS on H9c2 cells
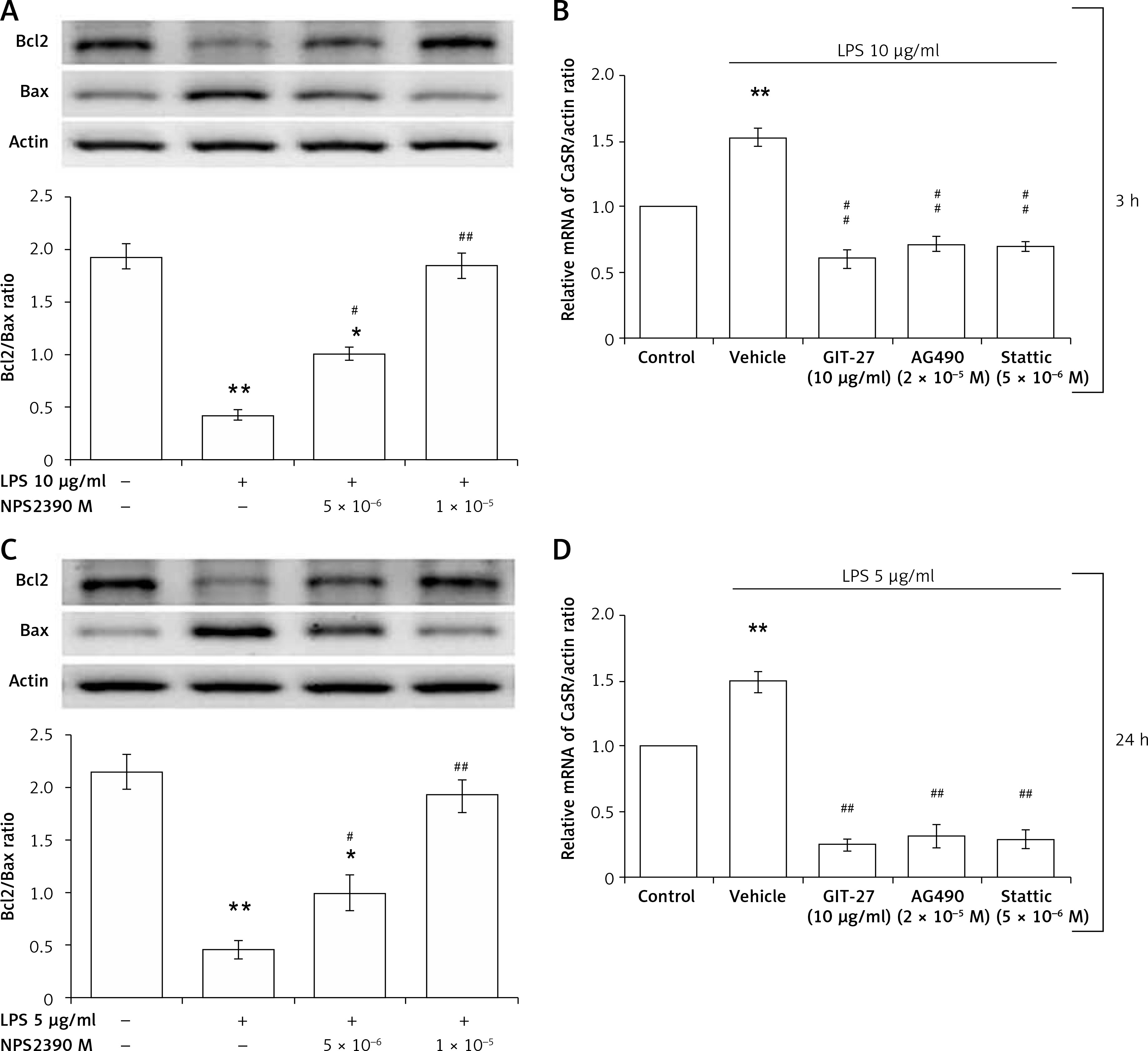
CaSR is believed to regulate cardiac function. Therefore, we used NPS2390, a CaSR-specific inhibitor, to investigate the potential role of CaSR in the toxic effects of LPS.
A 3-h incubation with 10 μg/ml LPS can inhibit Bax and Bcl-2 expression levels in H9c2 cells. This effect was reversed by NPS2390 in a dose-dependent manner (Figure 5 A). Additionally, the apoptotic signals observed in Western blots were also reversed by GIT-27, AG490, and Stattic treatment (Figure 5 B). Moreover, the hypertrophic signals attenuated by 24 h of 5 μg/ml LPS exposure were also alleviated by NPS2390 at the same dose (Figure 5 C). Similarly, the apoptotic signals detected by Western blots were attenuated by GIT-27, AG490, and Stattic treatment (Figure 5 D). These findings indicate that the toxic effects of LPS were associated with CaSR in H9c2 cells.
Figure 5
Effects of specific inhibitors on the changes induced by LPS. NPS2390 at the dose required to block the calcium-sensing receptor (CaSR) may reverse the apoptosis induced by LPS at a high dose after a short incubation (A) or at a low dose after longer treatment (C). CaSR mRNA levels were increased by LPS treatment after either a short incubation time at a high dose (B) or a longer incubation time at a low dose (D)
*P < 0.05 and **p < 0.01 vs. the vehicle-treated control (first column). #P < 0.05 and ##P < 0.01 vs. the low dose LPS-treated group (second column).
We then evaluated CaSR gene expression in H9c2 cells. CaSR mRNA levels were increased by LPS in a dose-dependent manner and plateaued in cells incubated with LPS at a high dose for a short exposure time (10 μg/ml for 3 h) and at a low dose for a longer exposure time (5 μg/ml for 24 h). As shown in Table II, the inhibitors specific to each signal in the TLR4 pathway can attenuate the effects of LPS in the same manner. Therefore, we found that LPS activated TLR4 to promote CaSR expression.
Table II
The effects of NPS2390 on LPS-modified hypertrophic biomarkers in H9c2 cells after a short (3 h) or longer (24 h) treatment time
| Incubation for 3 h | Control | LPS (10 μg/ml) | LPS (10 μg/ml) + NPS2390 (5 × 10–6M) | LPS (10 μg/ml) + NPS2390 (10–5M) |
|---|---|---|---|---|
| Relative mRNA of ANP/actin | 1.00 ±0.00 | 0.73 ±0.05** | 0.91 ±0.04*# | 1.10 ±0.04## |
| Relative mRNA of BNP/actin | 1.00 ±0.00 | v 0.69 ±0.04** | 0.86 ±0.04*# | 1.15 ±0.07## |
| Relative mRNA of β-MHC/actin | 1.00 ±0.00 | 0.67 ±0.032** | 0.92 ±0.06*# | 1.06 ±0.03## |
| Incubation for 24 h | Control | LPS (5 μg/ml) | LPS (5 μg/ml) + NPS2390 (5 × 10–6M) | LPS (5 μg/ml) + NPS2390 (10–5M) |
| Relative mRNA of ANP/actin | 1.00 ±0.00 | 0.56 ±0.03** | 0.81 ±0.04*# | 1.09 ±0.09## |
| Relative mRNA of BNP/actin | 1.00 ±0.00 | 0.42 ±0.03** | 0.76 ±0.05*# | 1.06 ±0.06## |
| Relative mRNA of β-MHC/actin | 1.00 ±0.00 | 0.53 ±0.05** | 0.81 ±0.02*# | 1.04 ±0.04## |
Discussion
In the present study, we demonstrated that the direct effect of LPS on isolated rat ventricular myocytes is markedly different from that on cardiac myocytes. Additionally, the effects of LPS on cardiac myocytes are associated with the treatment dose and related to the exposure time. The toxic effects of LPS on cardiac cells appear to be mainly mediated through proinflammatory cytokines and CaSR.
The present study focused on the effects of LPS alone, which are distinct from the changes that occur in animals or in clinical cases of sepsis; in those cases, the activation of endogenous inflammatory factors including cytokines and nitric oxide synthase 2 (NOS2) are involved [20]. The current study compared the changes after a 3-h exposure with those after a 24-h incubation. Among the acute effects after a 3-h exposure, low-dose LPS exposure activated hypertrophic signals in H9c2 cells in a dose-dependent manner, although apoptosis was also observed in cells exposed to the high dose of LPS. However, in H9c2 cells, apoptosis was only observed after a 24-h exposure to the low dose of LPS. This finding suggests a novel view that LPS activates hypertrophic signals prior to apoptotic signals in cardiac cells. The current finding is similar to the damage induced in cardiac myocytes by oxidants such as H2O2 [21] or potassium bromate [22], which induces cardiac hypertrophy at low doses but apoptosis at higher doses in vitro.
In ventricular myocytes isolated from adult rats, perfusion with LPS induces cardiac stimulation, which has not been mentioned before, although it diverges from cardiac depression after LPS treatment [23]. Cardiac cells exposed directly to LPS and cytokines in vitro have shown decreased [24–28], unchanged [8], and increased [26, 29, 30] contractility. Possible reasons for these divergent results may be associated with the experimental conditions used. Some studies investigated the effects using exposure to LPS or inflammatory mediators at various times such as 5 min [25] and 4 h [8]. The results regarding the toxic effects of LPS are mostly obtained from acute influences. The exposure times appear insufficient to activate downstream enzymes, including nitric oxide synthase 2 (NOS2) [20], and may not accurately replicate the effects observed in vivo. Furthermore, LPS initiates a sequence of cellular events that leads to reversible or irreversible injury in cardiac myocytes [31, 32]. Impairment of intracellular calcium homeostasis, alterations in excitation-contraction coupling, and enhanced programmed cell death are thought to be involved [33, 34]. Therefore, the effects of LPS on cardiac myocytes seem variable [7].
In research focused on cardiac myocytes, primary rat neonatal cardiomyocytes and the rat-derived H9c2 cell line are widely applied because both are similar in vitro [33]. We used H9c2 cells in the present study to establish the signalling pathway regarding TLR4 coupled with JAK/STAT3 for LPS, which has not been mentioned before. Pharmacological inhibitors, such as GIT27 for TLR4 [35], Tyrphostin AG490 for JAK [36], and Stattic for STAT3 [37], were also characterised in H9c2 cells. Thus, these inhibitors could be used in research by others in the future. Although it has been documented that LPS may rapidly induce STAT3 phosphorylation in cardiac myocytes [38], we failed to repeat this finding and instead observed all the signals within 30 min.
LPS-induced cardiac impairment has been demonstrated primarily via TLR4 [22]. CaSR activation is involved in endotoxin rat myocardial cell injury and apoptosis induced by LPS [39]. Using the Langendorff system, hearts isolated from CaSR knock-out (KO) animals showed a significantly lower basal heart rate than that in wild-type animals [40]. Moreover, heart dysfunction was also observed in TLR4-deficient hearts [41]. In animals, LPS-induced cardiac hypertrophy has also been demonstrated [42] after the binding with TLR4 [22]. It would be interesting to find out why apoptosis is observed in H9c2 cells only after a 24-h exposure to LPS, even at the low dose. The inhibitory effects of LPS on cardiac function are mainly mediated by the production of proinflammatory cytokines, such as IL-6 and TNF-α [43, 44]. Paracrine actions from cardiac myocytes have been reported to play an important role in the production of proinflammatory cytokines, such as IL-6 and TNF-α [45]. In the present study, IL-6 and TNF-α mRNA levels were elevated with increased time of exposure to LPS. Cardiac injury also progressed in the same manner. It seems possible that cardiac injury induced by LPS is associated with inflammation. Moreover, TLR4-mediated activation of STAT3 increases IL-6 production, which induces secondary STAT3 activation through the IL-6/gp130/JAK2 pathway [46].
In the clinic, cytokine activation has been introduced as one of the mechanisms for the progression of heart failure [47]. Recently, pyroptosis has been acknowledged as one type of programmed cell death [48]. LPS may induce common pathways (such as free radical production) to activate the NLRP3 inflammasome and then trigger caspase-1-dependent pyroptosis [49]. Inflammasomes are intracellular multiprotein complexes with caspase-1-activating platforms that can be activated by LPS [50], and the role of the inflammasome in sepsis has been documented [51]. Therefore, LPS activates NLRP3 inflammasomes to aggravate H9C2 cells as stimuli for pyroptosis and the inflammatory response [52]. Similarly, we applied BAY11-7082 to inhibit the inflammasome. The progression of cardiac injury by LPS in H9c2 cells was reduced by BAY11-7082, indicating the mediation of the inflammasome and/or pyroptosis. This new finding is a suitable explanation of why the effect of LPS progressed with time in H9c2 cells.
In the present study, we found that LPS promotes CaSR expression in H9c2 cells. CaSR is a member of the C subfamily of membrane G-protein coupled receptors, which is expressed in organs principally regulating systemic calcium homeostasis [53]. CaSR was involved in myocardial hypertrophy [54, 55] and heart failure [56]. CaSR is implicated in LPS-induced apoptosis through its increased expression, consequent Ca2+ overload, and TNF-α and IL-6 release [57]. In the present study, in contrast to the changes in expressions of proinflammatory cytokines, inhibition of the inflammasome did not markedly modify the CaSR expression in H9c2 cells receiving a long exposure of LPS. Therefore, LPS might have another way to modulate CaSR expression. However, further evidence to support this hypothesis is required. Moreover, both hypertrophy and apoptosis in H9c2 cells due to LPS were diminished by NPS2390 at a dose sufficient to inhibit CaSR [58]. The reversal of apoptosis due to acute treatment with LPS by NPS2390 was also observed in cardiac myocytes from neonatal rats [39]. Interestingly, NPS2390 inhibited the hypertrophic response to LPS and the apoptosis induced by LPS at same dose. It seems that the role of CaSR in LPS-induced cardiac injury is not simple, and a detailed investigation is required. Additionally, CaSR has been reported to play a role in promoting cardiac fibrosis [59]. Pharmacological inhibitors specific to each signal in the TLR4-coupled JAK/STAT pathway blocked the effects of LPS on CaSR expression, which may enhance the potential role of the TLR4-coupled JAK/STAT pathway in CaSR activation.
In the clinic, LPS from the outer membrane of bacteria is the primary trigger of the systemic inflammatory response in sepsis [1]. Sepsis or septic shock occurs worldwide, with nearly half of these patients dying each year [60]. The clinical manifestations of sepsis are largely attributable to the LPS-induced changes in both myeloid cells (macrophages) and non-myeloid cells (endothelial cells and cardiomyocytes). These events are the primary cause of myocardial dysfunction in sepsis, which is an important determinant of patient outcome. In the heart, LPS exerts its effects in two overlapping phases. Reduced systolic function and contractile reserve occur within minutes to hours of LPS exposure [61]. Early myocardial dysfunction has been related to direct LPS effects and the rapid LPS-stimulated production of proinflammatory cytokines. Proinflammatory cytokines and other mediator responses to LPS result in injury from a variety of mechanisms, including free radical production, nitric oxide generation, and arachidonic acid metabolite release. These events result in progressive contractile dysfunction, including heart failure [62]. Chronic heart failure appears to be accompanied by a persistent increase in inflammatory cytokines. Recently, LPS-induced hypomyelination has also been demonstrated in forebrain slices of neonatal rats [63]. However, CaSR is ubiquitously expressed in the human body where it activates multiple signalling pathways, and the role of CaSR in the heart is still quite controversial [64]. Few reports have been conducted the application of CaSR-related agent(s) in clinical practice.
The present study has some limitations, as follows. In LPS-treated Langendorff hearts, the changes in contractility, the expressions of TLR4/JAK2/STAT3/CaSR and molecules involved in apoptosis or hypertrophy should be studied further. The effects of LPS in the hearts isolated from TLR4 or CaSR deficient animals should also be investigated in advance. Moreover, it has been documented that IL-6 trans-signalling modulates TLR4-dependent inflammatory responses via STAT3 [65]. Further research including the crosstalk among JAK/STAT-, TLR-, and LPS-related pathways in cardiomyocyte should be investigated in our future work. Additionally, applications of GIT27, AG490, Stattic, or IL-6 blockade to investigate the changes of Ca2+ mobilisation and apoptosis in LPS-treated hearts should also be performed in advance.
In conclusion, we are the first to demonstrate the direct effects of LPS on isolated ventricular myocytes mounted on a Langendorff apparatus. In contrast to the inotropic effects in isolated ventricular myocytes, the toxicity induced by direct LPS treatment in cardiac myocytes was observed to be both time- and dose-dependent. The influence of LPS on cardiac cells is mainly induced by the activation of the TLR4-coupled JAK/STAT pathway. We also found that LPS activates hypertrophic signals prior to apoptotic signals in cardiac cells. The time-dependent changes by LPS are associated with cytokines, although the inflammasome and LPS dosage are also involved. Moreover, CaSR activation also mediates cardiac injury induced by LPS. However, CaSR expression was not markedly reduced by inflammasome inhibitor as the progression of inflammation in H9c2 cells. Otherwise, CaSR is not a good target for the alleviation of LPS-induced injury because CaSR is ubiquitously expressed in the human body. Thus, inhibiting the inflammasome and/or inflammation would be helpful in the future.