
Introduction
Chronic pain lasts for 3 months or longer and is experienced by up to 30% of adult populations [1] and 42% of the elderly aged 65 years and over [2]. Understanding the neurobiological mechanisms underlying pain is essential to improve treatments for chronic pain. Previous pain studies have mainly focused on the role of the spinal dorsal horn [3–5]. However, imaging studies have shown that the anterior cingulate cortex (ACC) is activated in patients with acute or chronic pain [6–8], which is further supported by the findings from independent studies that employed different techniques such as intracranial electrical stimulation, pharmacology, and induced lesions that consistently implicate the important role played by the ACC in the regulation of pain [9–11]. However, the technical limitations of the above methods make it challenging to study how ACC integrates into the neural pain circuitry. Recently, an increasing number of studies have demonstrated the important regulatory role of the ACC in inflammatory- [12] and cancer-induced pain [13] in freely moving mice using optogenetic manipulation of neuronal activity.
A previous study reported that chronic pain-related central sensitization induces a widespread alteration in the activity of the somatosensory network, which is mainly mediated by the ACC and the striatum [14]. Furthermore, studies have also shown that dopamine in the ventral striatum is involved in pain and reward events in the human brain [15, 16]. Functional imaging (e.g. fMRI) studies show that the ventral striatal neurons are activated by noxious stimuli including cutaneous electrical stimulation [17]. Pharmacological regulation of the striatal dopamine levels also alters pain and reward-related behavior [18, 19]. Altogether, these studies suggest that the striatum is important for the perception of pain. A major ACC projection target is the dorsomedial striatum (DMS) [20]. Neurons in the ACC project to the DMS as part of a corticostriatal circuit with putative roles in learning and other cognitive functions [21]. However, the exact function of this corticostriatal circuit in pain regulation is poorly understood.
Optogenetics has been widely used in neuroscience research to selectively and precisely control the activity of a defined group of central neurons to determine their roles in behavioral functions in animals [22]. It is an established and reliable methodology for fast activation or inhibition of primary neuronal activities by light [23] . The most critical opsins are the blue light-sensitive cation channel channelrhodopsin-2 (ChR2) and the yellow light-sensitive Natronomonas pharaonis halorhodopsin (NpHR) [24]. Under the blue light of 473 nm, the ChR2 light-sensitive channel can be activated rapidly, causing extracellular cation influx and depolarization of cells [25]. Yellow light activates the NpHR light-sensitive channel protein, causing extracellular chloride influx, which leads to cell hyperpolarization [26]. The excitatory neuron-specific promoter calcium-calmodulin dependent protein kinase IIα (CaMKIIα) is specifically active in glutamatergic neurons to drive reporter gene expression in the neurons [27]. Optogenetics has been used in recent studies of pain [28, 29].
Although the above studies have demonstrated the important roles of ACC excitatory neurons in chronic pain, other questions remain unanswered. Thus, this study sought to determine whether the known axonal projections from the ACC to the dopaminergic neuron-containing dorsomedial striatum (DMS; the ACC-DMS projection) regulate pain, and specifically neuropathic pain in the chronic constriction injury (CCI) mouse model.
Material and methods
Experimental animals and groups
Adult male C57BL/6 mice (6 to 8-week old; Slack Animal Center, Shanghai Institute of Bioscience and Biotechnology) were used in this study. All procedures in this study were approved by the WenZhou Medical University Institutional Animal Care and Use Committee to ensure minimal animal use and discomfort (Number: wydw2016-0100). Mice were kept on a 12 : 12 light-dark cycle with food and water provided ad libitum. Animals were given an average of 7 days to adjust to the new environment before beginning the experiment.
For ACC: the viral vectors were unilaterally injected into the left ACC, and the cannulas were implanted into the left ACC. The ChR2-on-left group was injected with AAV2/9-CaMKIIα-ChR2-mCherry, stimulated by the blue light (on) and the left planter was measured. There were four group: ChR2-off, ChR2-on, NpHR-off and NpHR-on. The left and right hind limb mechanical and thermal pain thresholds were measured.
For ACC-DMS: the viral vectors were unilaterally injected into the left ACC, and the cannulas were implanted into the left DMS. The NpHR-off-right group was injected with AAV2/9-CaMKIIα-eNpHR3.0-GFP, without stimulation by the yellow light (off) and the right planter was measured. There were six group: ChR2-off, ChR2-on, NpHR-off, NpHR-on, mCherry-off and mCherry-on. The left and right hind limb mechanical and thermal pain thresholds were measured.
Optogenetics and viral vectors
In order to achieve specific ChR2 or the third NpHR (eNpHR3.0) expression in the central nervous system in mice, we used adeno-associated virus (AAV)2/9 as a vector [30] and the calcium-calmodulin dependent protein kinase IIα (CaMKIIα) as a promoter, which was fused to mCherry or green fluorescent protein (GFP). The recombinant virus AAV2/9-CaMKIIα-ChR2-mCherry (1.1 × 1012 µg/ml), AAV2/9-CaMKIIα-eNpHR3.0-GFP (1.5 × 1012 µg/ml) and their control (AAV2/9-CaMKIIα-mCherry (1.3 × 1012 µg/ml)) were purchased from Hanbio Biotechnology Company (Shanghai, China).
Stereotaxic viral injections and cannula implantation
Mice were anesthetized with isoflurane (1.5–2%) as previously described [31]. In all experiments, the viral vectors were unilaterally injected into the ACC only using a 33Ga, 10-µl Hamilton syringe (0.5 µl over 10 min). For ACC, the injection site coordinates were anteroposterior (AP) +0.98 mm from bregma, mediolateral (ML) +0.32 mm, and dorsoventral (DV) –1.6 mm (zeroed at the dura), according to the atlas of George Paxinos of the Mouse Brain in Stereotaxic Coordinates, second edition. The needle was held in place for 10 min after the injection ended to allow for diffusion with minimal spread of viral particles along the injection track. Mice were then unilaterally implanted with an optic cannula (PlasticsOne) in the ACC (AP +0.98 mm, ML +0.32 mm, DV –1.4 mm). For DMS, mice were stereotaxically implanted with an optic cannula: AP +1.00 mm, ML +1.30 mm, DV –2.6 mm. The cannulas were fixed to the skull with anchoring screws and acrylic cement. The 200 µm core optical fibers were threaded through guide cannulas (Figures 1 A, 2 A, B).
Figure 1
Viral vector expression in the anterior cingulate cortex (ACC). A – The expression of AAV2/9-CamKII-ChR2-mCherry (blue-sensitive, 473 nm) and AAV2/9-CamKIIeNpHr3.0-GFP (yellow-sensitive, 589 nm) (100×). Scale bars = 100 μm. B – The expression of c-fos in the brain is activated by the 10-min-blue light (473 nm) stimulation in the ACC of the ChR2 and mCherry groups (400×). Scale bars = 50 μm. C – The expression of c-fos in the ChR2 group is significantly higher than that of the mCherry group (n = 3, *p < 0.05). The arrows represent c-fos
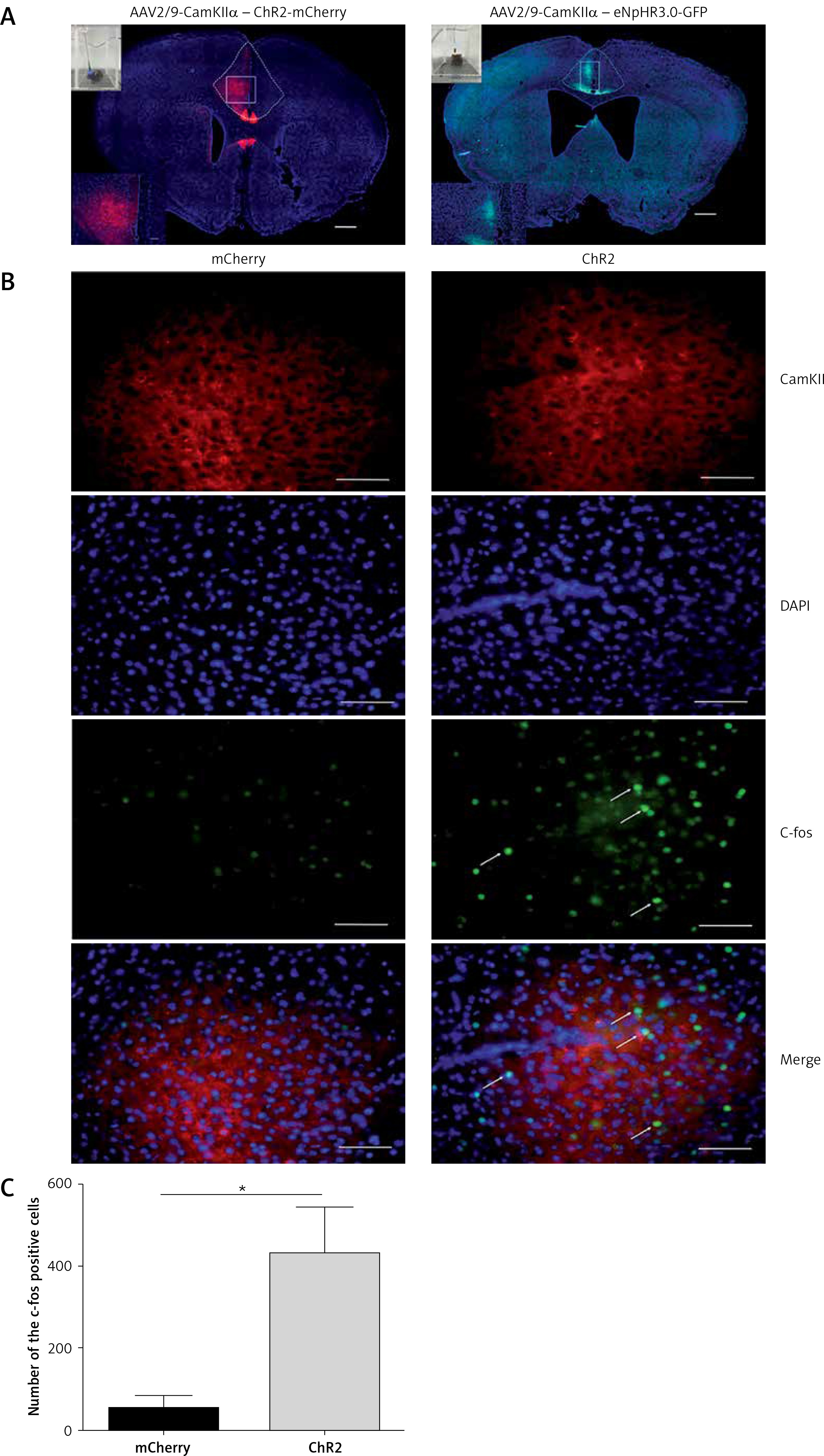
Figure 2
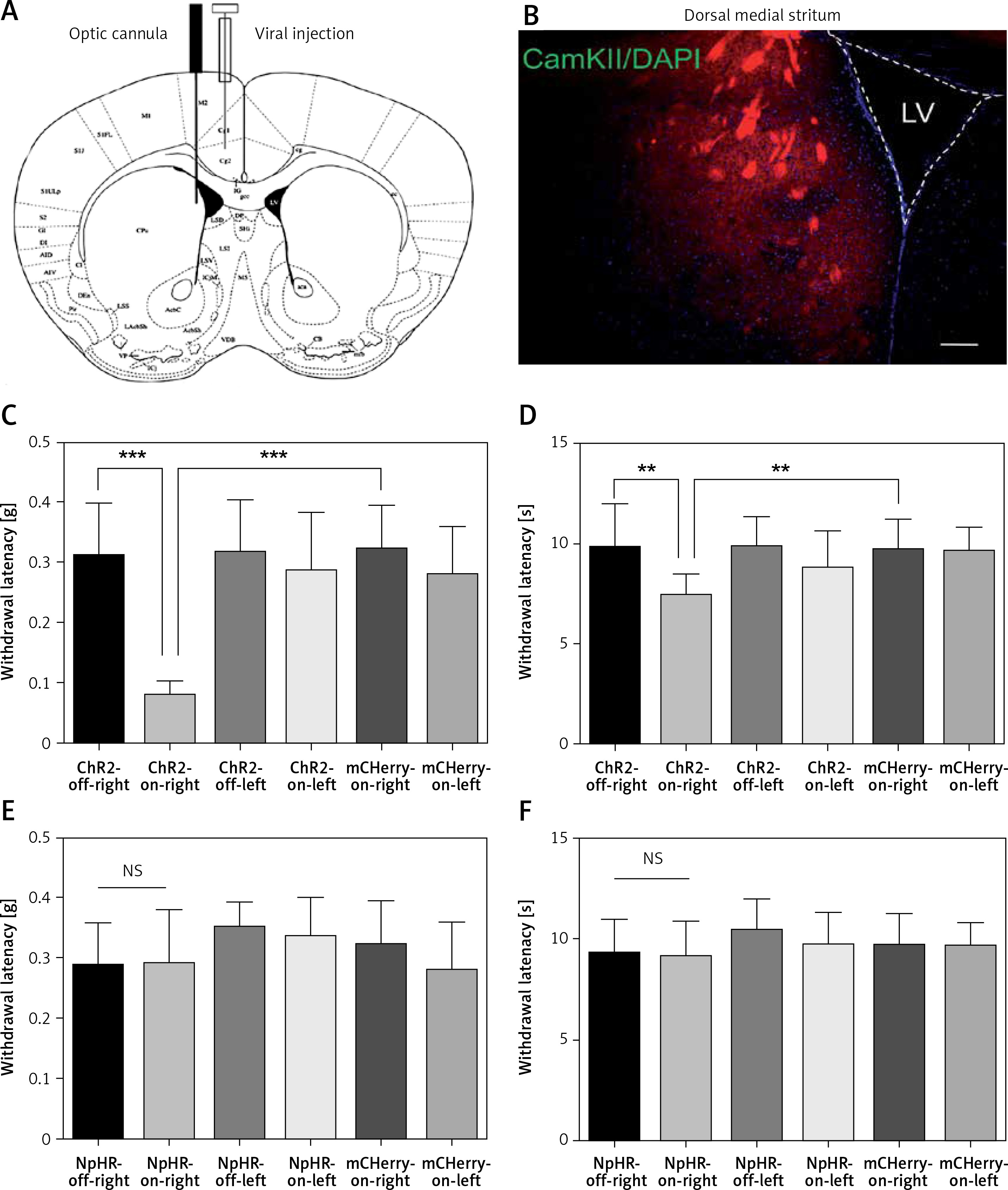
Activation of anterior cingulate cortex (ACC) excitatory projections in the dorsomedial striatum (DMS) reduces the pain threshold in normal mice. A – Injection and implantation locations. B – The ACC neurons projecting downstream to the DMS (100×). Scale bars = 100 μm. C, D – are the mechanical pain and thermal pain threshold tests when blue light stimulated the DMS of the normal mice (n = 10 or 12; *p < 0.05, **p < 0.01, ***p < 0.001). E, F – show the mechanical pain threshold test and thermal pain threshold test for normal mice when the yellow light was applied to the DMS (n = 9; p > 0.05)
Optical stimulation
The 200 µm optic fibers in the cannulas were linked to the fiber optic rotary joint, and then connected to a 473-nm blue or 589-nm yellow laser (NEWDOON, Hangzhou Newdoon Technology Co., Ltd). The blue light stimulation parameters were 10 Hz, 10 nm wave width, and tip light intensity of 5 mw/mm2, whereas the yellow light stimulation was 10 Hz, wave width 50 nm, and tip light intensity 10 mw/mm2 [12, 13]. The light stimuli were used at the same time during the behavioral test. The light source was turned off at the end of one color test, and a 5-min rest interval was allowed before the next color test. After the behavioral test, mice were sacrificed by perfusion and the optical fiber position was verified for each animal. Only mice that had accurate optical fiber positions were chosen for analysis.
Chronic constriction injury (CCI)
After 14 days of the virus injection and fiber implantation, the basic pain behavior test was performed, and the day of establishing the neuropathic pain model was recorded as day 0. CCI was induced as previously described [32]. Briefly, mice were deeply anesthetized with 1.25–1.5% isoflurane and the right sciatic nerve was exposed at the mid-thigh level. After removal of adherent tissue, three loose ligatures of chromic gut 6/0 (Jinhuan Medical Product Co., Shanghai, China) were tied 1 mm apart around the nerve. Muscle and skin were closed with sutures (Silkam 6/0 and Safil 4/0, respectively; Ethicon). Pain behavior was tested on the third and seventh days after CCI.
Mechanical allodynia test
Mechanical sensitivity was assessed using the up-down method with von Frey filaments as previously described [33]. Mice were acclimated on a wire-mesh floor in Plexiglas cubicles for 1 h, after which mechanical sensitivity was evaluated using a set of calibrated von Frey fibers (0.008, 0.02, 0.04, 0.07, 0.16, 0.4, 0.6, 1.0, 1.6, and 2.0 g; Stoelting Co., Wood Dale, IL USA) applied to the plantar surface of the hind paw for a few seconds until the fiber bent slightly. The appearance of any of the following behaviors was considered as a withdrawal response: (1) rapid flinch or withdrawal of the paw, (2) spreading of the toes, or (3) immediate licking of the paw. If the animal moved the paw for some other reasons before the end of the 2 s, the test was considered ambiguous and repeated. If a positive response to the first stimulus was obtained (i.e. the 0.16-g filament), then the next, smaller filament was applied; if there was no response, the next larger filament was used. After the first positive response, the experiment was continued until four additional responses were obtained. Stimuli were applied with 5 min time intervals, and the 50% paw withdrawal threshold value was calculated.
Thermal pain test
Thermal allodynia was assessed with a Hargreaves apparatus as previously described [34]. The heat source (390-Plantar Test, IITC Life Science) was placed with the guide light pointing toward the plantar surface of the hind paw and the thermal beam was turned on. A paw withdrawal stopped the test, at which point the withdrawal latency was recorded. Minimum and maximum cut-offs were assigned to 1 and 20 s, respectively. The measurements were repeated five times with 5 min intervals for each paw, and then averaged and analyzed. The result is expressed as the mean withdrawal latency for each animal and group ± standard error of the mean (SEM).
Immunofluorescence
Mice were deeply anesthetized with isoflurane, then intracardially perfused with perfusion buffer followed by 4% paraformaldehyde (PFA) in phosphate buffer (PB), pH 7.4, at room temperature for 30 min. Brains were fixed in 4% PFA overnight at 4°C and transferred to 30% sucrose in PBS to equilibrate for 3 days. Then, 30 µm coronal sections were cut on a cryostat (Leica) and washed with PBS for 10 min. Sections were washed with PBS with 0.3% Triton X-100 (PBST) for 10 min, and incubated in PBST containing 3% normal donkey serum (NDS) for 1 h at room temperature. Sections were incubated with primary antibody, anti-c-fos (1 : 500; Abcam) diluted in PBST with 3% NDS overnight at 4°C. Sections were then washed in PBS and incubated for 2 h at room temperature with Alexa Fluor 488 (1 : 500; Invitrogen) and Alexa Fluor 594 (1 : 500; Invitrogen) in 5% normal goat serum and NDS in PBST. Sections were washed in PBS and coverslipped with Vectashield mounting medium with DAPI. Slides were stored at 4°C until use.
Statistical analysis
Quantitative analysis of immunofluorescence staining was performed using the ImageJ professional image analysis system. All statistics were run on IBM SPSS Statistics 13.0 software. The data are expressed as mean ± SEM; comparisons between two groups were performed using Student’s t-test, and comparisons between three groups were performed using ANOVA with a post hoc Bonferroni’s test for further comparison. Comparisons between different time points were made using repeated measures ANOVA. The variance homogeneity test was performed with Levene’s test. Statistical significance was set at p < 0.05.
Results
Virus expression in the ACC
We used optogenetics to selectively and temporarily activate or silence ACC excitatory neurons to study the role of ACC in pain modulation. First, we injected the vector into the ACC and implanted the fiber sleeving. Two weeks following the vector injection, the expression of the virus was restricted to the ACC (Figure 1 A). To ensure that ChR2 was functional in the brain, we examined the expression of c-fos immunoreactivity, a marker for neuronal excitation. Mice were divided into two groups: the ChR2 group (AAV2/9-CaMKIIα-ChR2-mCherry) and the control mCherry group (AAV2/9-CaMKIIα-mCherry) and were exposed to blue light (473 nm). Compared with the mCherry group, the expression of c-fos in the ACC region of the ChR2 group was significantly increased (n = 3, *p < 0.05, Figures 1 B, C). These data demonstrated that blue light stimulation of the ACC region activated putative excitatory neurons.
Activation of ACC excitatory neurons reduces pain threshold in normal mice
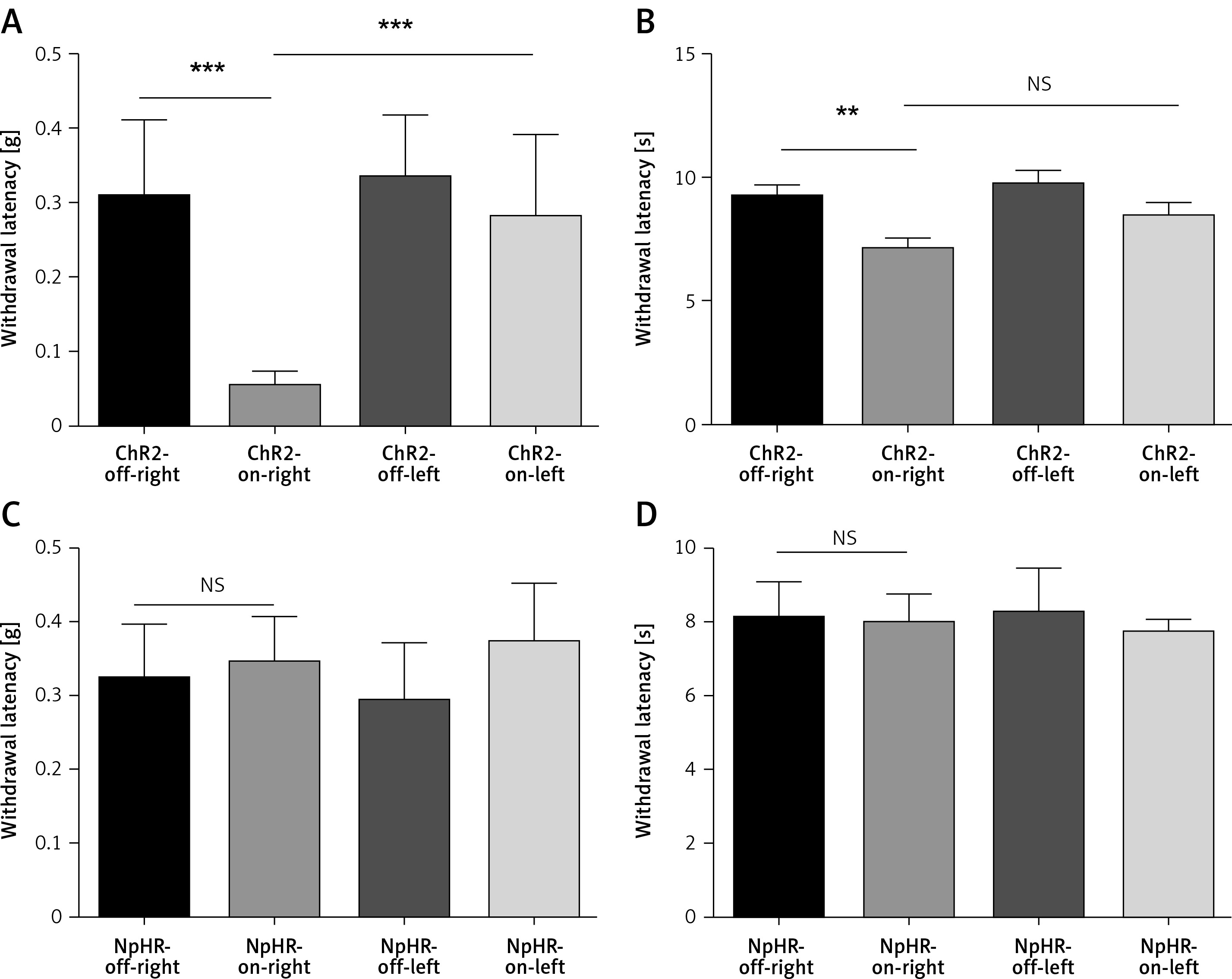
We found unilateral photoactivation of the left ACC-induced mechanical and thermal allodynia, as shown by a decrease in the mechanical threshold (Figure 3 A) and reduced heat sensitivity latency (Figure 3 B) in the ChR2-on-right (n = 12) group compared to the ChR2-off-right group of the normal mice (n = 10, **p < 0.01, ***p < 0.001). Additionally, mechanical and heat pain thresholds of the left foot were tested (Figures 3 A, B) during unilateral stimulation of ACC excitatory neurons, which showed an obvious change in the mechanical pain thresholds (Figure 3 A). However, when the ACC pyramidal excitatory neurons were inhibited, there were no significant changes in the mechanical and heat pain thresholds of the foot (Figures 3 C, D). Thus, the right thigh was chosen as the chronic constriction injury of the sciatic nerve model for the following experiments.
Figure 3
Activation of anterior cingulate cortex (ACC) excitatory neurons leads to hyperalgesia in normal mice. A, B – show the mechanical pain and thermal pain threshold test results between the ChR2-off-right group (n = 10) and the ChR2-on-right group (n = 12, **p < 0.01, ***p < 0.001). C, D – show the mechanical pain and thermal pain threshold test results between the NpHR2-off-right group and the NpHR-on-right group (n = 7, p > 0.05)
Inhibition of ACC excitatory neurons relieves neuropathic pain
Next, ACC excitatory neurons that expressed eNpHR3.0-GFP in the CCI mice were inhibited using the yellow light. The mechanical pain threshold and thermal pain latency of the mice with sciatic nerve injury were measured on the 3rd and 7th day following sciatic nerve ligation. Compared with the ChR2-off-right group (n = 8), the ChR2-on-right group (n = 10) demonstrated no significant difference in mechanical pain (Figure 4 A) or thermal pain thresholds (Figure 4 B) upon blue light activation of ACC excitatory neurons. Illumination with yellow light that inhibited the ACC resulted in a significant improvement in the NpHR-on-right group (n = 10) in mechanical pain threshold (Figure 4 C) and heat pain latency (Figure 4 D) compared to the NpHR-off-right group (n = 9, *p < 0.05, ***p < 0.001).
Figure 4
Inhibition of excitatory neurons in the anterior cingulate cortex (ACC) relieves hyperalgesia in the CCI mouse model. A, B – show the mechanical and thermal pain threshold test results of CCI mice when stimulated with blue light in the ACC (n = 8 or 10, p > 0.05). C, D – show the mechanical pain threshold test and thermal pain threshold test results of CCI mice when the ACC was inhibited by yellow light (n = 9 or 10, *p < 0.05, ***p < 0.001)
Activation of neurons in the ACC-DMS reduces pain threshold in normal mice
Immunofluorescence staining was performed 6 weeks after the virus injection in the ACC. Expression of the virus was also detected in the DMS. Then, the AAV2/9-CamKII-ChR2-mCherry, AAV2/9-CamKII-eNpHR3.0-GFP, or AAV2/9-CamKII-mCherry virus was injected in the left ACC and an optic fiber was implanted into the left DMS (Figure 2 A). Expression of the virus was observed in fibers in the DMS projecting from the ACC (Figure 2 B). The mechanical pain threshold and the heat latency in the ChR2-on-right group (n = 12) were significantly lower than those of the ChR2-off-right group (n = 10) (Figures 2 C, D). Unilateral stimulation of the DMS with yellow light did not alter the mechanical pain threshold (Figure 2 E) or thermal pain latency (Figure 2 F) in the NpHR-off-right group or NpHR-on-right group (n = 9).
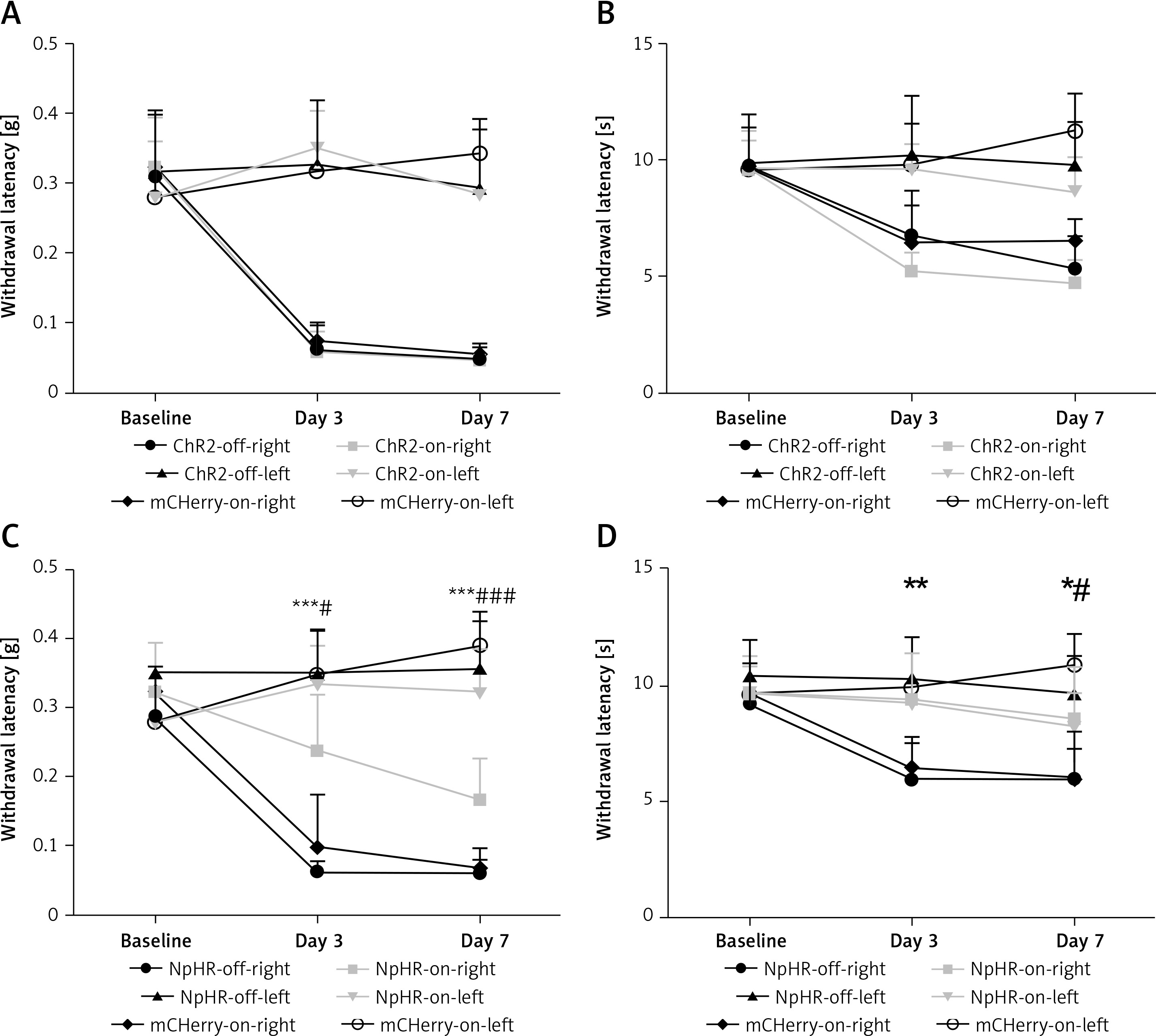
Inhibition of the ACC-DMS relieves the effect of neuropathic pain
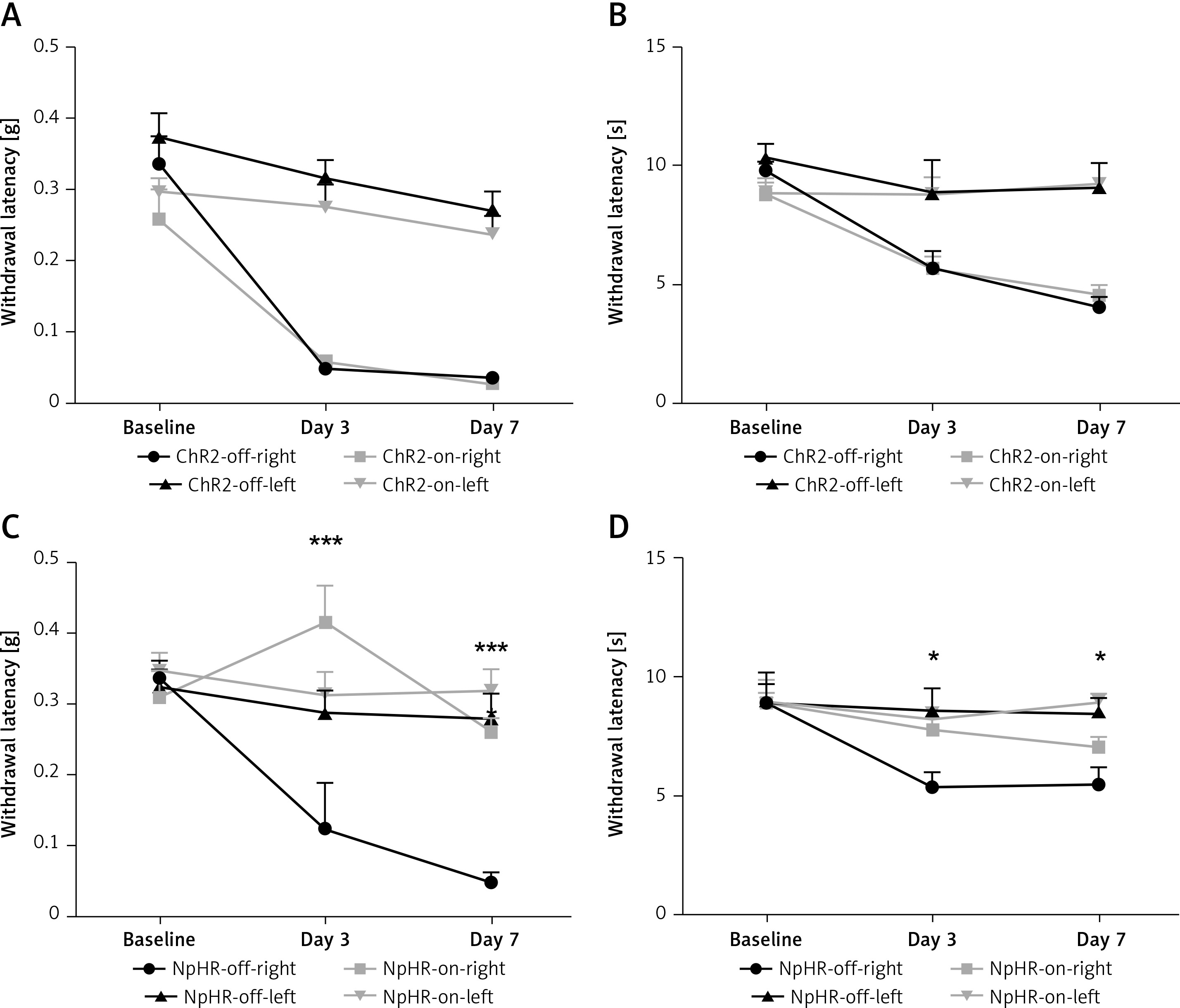
We inhibited the DMS of mice injected with eNpHR3.0 or mCherry virus in the ACC after sciatic nerve ligation. We found that the mechanical pain threshold (Figure 5 A) and thermal pain threshold (Figure 5 B) in mice of the ChR2-on-right group (n = 10) were not significantly different on day 3 or day 7 after CCI, compared with the ChR2-off-right (n = 8) group when the DMS was activated by the blue light. There was a significant improvement in the mechanical (Figure 5 C) and thermal pain thresholds (Figure 5 D) during unilateral stimulation with yellow light in the NpHR-on-right group (n = 9) compared with the NpHR-off-right group (n = 8) (*p < 0.05). Compared with the NpHR-on-left group, the mechanical pain threshold of the NpHR-on-right group was significantly different on day 3 (#p< 0.05) and day 7 (###p < 0.001), and the thermal pain threshold was improved on day 7 (#p < 0.05), indicating that inhibition of the DMS could relieve hyperalgesia in CCI mice (Figure 5 D).
Figure 5
Inhibition of the anterior cingulate cortex (ACC) projections to the dorsomedial striatum (DMS) relieves pain in CCI mice. A, B – show the mechanical pain and thermal pain threshold test results for CCI mice during blue light stimulation of the DMS region between the ChR2-off-right group and the ChR2-on-right group (n = 8 or 10, p > 0.05). C, D – show the mechanical pain thresholds and thermal pain thresholds for CCI mice during yellow light inhibition of the DMS on day 3 and day 7 in the NpHR-off group and NpHR-on group (n = 8 or 9, *p < 0.05, **p < 0.01, ***p < 0.001).
*NpHR-on-right groups versus NpHR-off-right group; #NpHR-on-right group versus NpHR-on-left group.
Discussion
In this study, normal male C57BL/6 mice and a CCI mouse model were selected, and on day 3 and day 7 following CCI, the tests showed that the mechanical pain threshold of the affected side was significantly decreased and the thermal pain latency was significantly shortened, indicating successful establishment of the model.
We observed that activation of the ACC excitatory neurons reduced the mechanical pain threshold in normal mice and shortened the latency of the response to heat-related pain, while inhibition of ACC excitatory neurons improved the pain threshold in neuropathic pain model mice, which are consistent with findings from previous studies [12, 13].
For neuropathic pain, there is excessive excitation and irregular firing of neurons. Peripheral nerve injury leads to enhanced NMDA activity, glial activation and hypertrophy within the spinal cord. In addition, activated microglia express purinergic receptor subtypes and release pro-nociceptive cytokines such as IL1, TNF-α, and neurotrophin factors, which exacerbate nociceptive transmission and ultimately maintain the symptoms of hypersensitization [35, 36]. Central sensitization is dominant in neuropathic pain. In fact, not only neurons but also glial cells (e.g. astrocytes and microglia) and infiltrating mast cells are involved in the generation and maintenance of central sensitization [37]. Central sensitization is also associated with the expansion of receptive fields of dorsal horn neurons, reduction of central inhibition, and persistent spontaneous dorsal horn neuron activity. Such activity results in a sensory response to low-intensity stimuli. Sensitization of the nervous system in response to neuropathy and chronic pain is caused by changes in neuronal structure, connections between neurons, and changes in the number and nature of neurotransmitters (e.g. GABAA), receptors (e.g. A2A, P2X3), and ion channels (e.g. ASIC) [38]. The adaptation of structural and functional, namely neural, plasticity, leads to a balance transition between excitatory and inhibitory systems, and ultimately results in increased pain [39].
Previous research using electrical stimulation and pharmacological methods confirmed the involvement of ACC in chronic pain [40]. Compared with pharmacologic and direct electrical stimulation, optogenetic technology provides higher temporal (ms level accuracy) and spatial accuracy, which are important in defining the causal relationships between neuronal activity and pain. Nonetheless, optogenetic technology also has its drawbacks. The use of light from a laser or LED light source produces heat, and prolonged light exposure may bring detrimental effects to the tested organisms, which renders it unsuitable for long-duration studies of neurobiological processes. In this study, we combined light stimulation with behavioral tests to confirm the role of ACC neurons in neuropathic pain.
Figures 3 A and B show that when excitatory neurons in the ACC region of normal mice were stimulated by the blue light, the mechanical pain threshold was more significant than the thermal threshold, which suggests that the ChR2 protein activated in the pyramidal neurons may cause Na+ influx and leads to depolarization of neurons, increases excitatory synaptic strength, and results in neurological mechanical hypersensitivity [41–43]. In addition, the pain signals caused by activating excitatory neurons in the ACC region can be transmitted to undamaged tissues, causing secondary hyperalgesia. Since the response of sensitized dorsal horn neurons is exaggerated relative to normal physiological conditions [44, 45], the mechanical pain and heat pain of the left and right foot of the normal mice illuminated have a downward trend, and the trend of mechanical pain is more obvious.
Recent studies have reported the role of multiple forebrain regions, such as the amygdala, ACC, and nucleus accumbens (NAc), in the development of chronic pain [46–48]. The NAc is functionally related to the motivation-reward circuitry, but has also recently been shown to play an important role in pain regulation [49–51]. In this study, we have demonstrated that the activation of the DMS through projections from ACC excitatory neurons had the same result as direct activation of ACC excitatory neurons (Figures 4 C, D). Transient receptor potential vanillin type 1 (TRPV1) is an important pain-related receptor and repeated morphine therapy has been shown to upregulate TRPV1 in the dorsal striatum [52], suggesting that the striatum is involved in the development of pain. In this study, we found that the neuronal endings of downstream projections of ACC excitatory neurons reduced the pain threshold in mice, whereas inhibition of the DMS nerve endings projecting from the ACC excitatory neurons could relieve neuropathic pain in mice (Figures 4 C, D), suggesting that the DMS may be involved in neuropathic pain.
Glutamate is the major excitatory neurotransmitter for ACC projecting neurons, and most of the neurons in the DMS are dopaminergic neurons. Studies have reported that midbrain dopaminergic neurons may co-release the excitatory neurotransmitter glutamate [53–55], and that metabotropic glutamate receptor subtype 8 (mGluR8s) in the DMS are likely to selectively modulate thermal pain induced by nerve injury [56], which raises the speculation that glutamate may act as a transmitter that facilitates nociceptive transmission in the ACC, thereby stimulating the striatal dopaminergic neurons to release glutamate, which results in corresponding behavioral changes.
The classic pathway of central nervous system regulation of chronic pain is mainly through the downstream inhibition pathway, i.e. pain activation of the periaqueductal gray (PAG) projection to the rostral ventral medulla (RVM), and then the pain signal transfers to the spinal dorsal horn [57–59]. Studies have shown that the ACC may project to the PAG [60]. Thus, it is possible that the ACC regulates hyperalgesia through the downregulation pathway. Previous studies have shown that stimulation of the rat ACC region causes a flick reaction that may be mediated by a brain stem downstream pathway [61]. Our data demonstrate that targeting ACC-DMS activities can attenuate pain symptoms in mice. Additionally, activation of the prefrontal cortex, especially the anterior marginal cortex, results in hyperalgesia relief [62], which can also be recapitulated by activating its downstream projection, the NAc [63]. This suggests that different corticostriatal circuits may have different regulatory roles in the development of pain.
The most important contribution of our study is the identification of a corticostriatal circuit in pain regulation. The ACC-DMS projection provides top-down modulation of histaminergic itch-related behavior [20]. Human imaging studies showing altered connections between the ACC and DMS in the chronic pain states emphasize the importance of the corticostriatal circuit [64]. Our behavioral studies indicate that this corticostriatal circuit can be a key mechanism for the brain to exert top-down control for pain.
Electrophysiological technology was not employed to verify virus activity in this study. Instead, immunofluorescence staining and quantification of the early-response protein c-fos was used to detect neuronal activation (Figures 1 B, C). One of the limitations of this study is that we did not distinguish between glutamatergic pyramidal neurons and GABAergic interneurons in the ACC by selective optogenetic targeting. However, a previous study demonstrated that the activation of ACC parvalbumin (PV)-expressing interneurons has an analgesic effect in inflammatory pain, whereas activation of the ACC somatostatin (SOM)-expressing interneurons has no effect on pain thresholds [65]. In the future, there will hopefully be a promoter-specific strategy that can directly dissect the pain-modulatory roles of pyramidal neurons versus interneurons.
In conclusion, this study confirms a novel corticocortical circuit in the regulation of chronic neuropathic pain. The projection from the ACC to the DMS provides important antinociceptive effects. Neuromodulation therapy for pain remains limited to spinal or peripheral nerve stimulation. Our findings suggest the potential of targeting the ACC-DMS circuit in the brain for treating intractable pain.