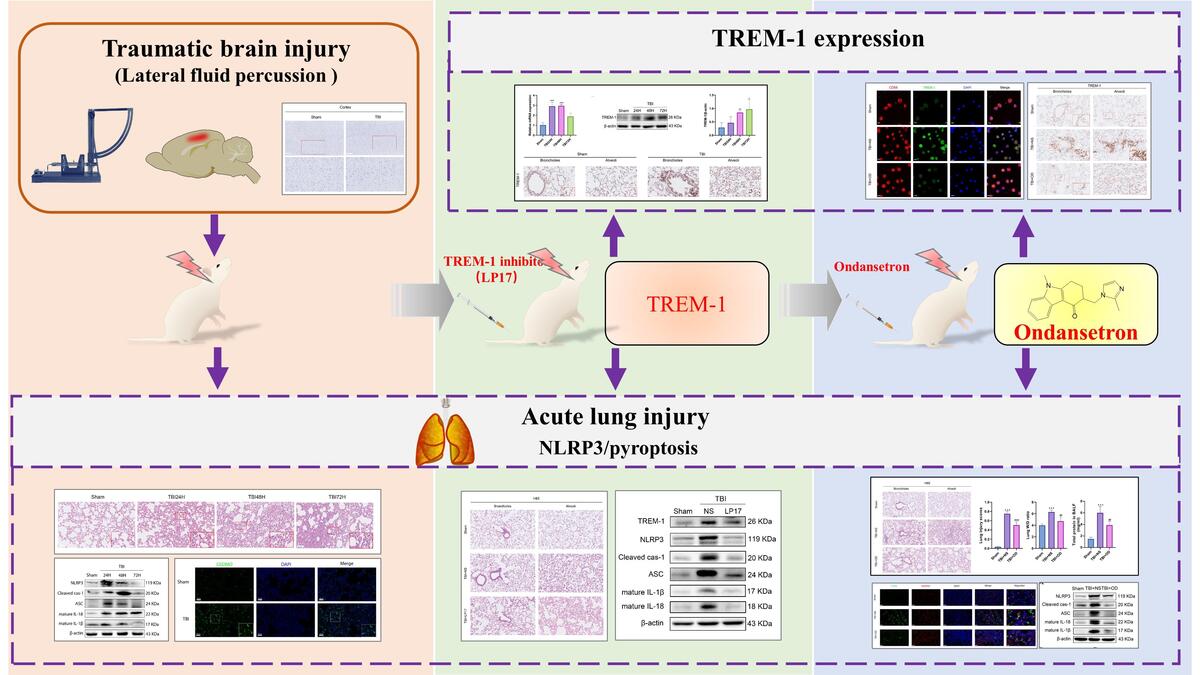
Introduction
Traumatic brain injury (TBI) is a critical public health concern with high mortality and morbidity worldwide [1]. Approximately 20–30% of TBI patients develop acute lung injury (ALI), and lung dysfunction further exacerbates TBI [2, 3]. To date, the pathogenesis of TBI-induced ALI (TBI-ALI) remains unclear.
Systemic inflammatory responses play a crucial role in the pathogenesis of TBI-ALI [4]. Pyroptosis is a type of cell death that possesses potential for inflammation and, at an increased rate, plays a critical role in promoting systemic inflammation. NLR family pyrin domain containing 3 (NLRP3) inflammasome activation is a key step in pyroptosis [5, 6]. It has been shown that TBI activates NLRP3 inflammation and pyroptosis in lung tissue and aggravate TBI-ALI [7]. Moreover, ameliorating inflammasome-induced pyroptosis could attenuate TBI-ALI [8]. After pro-caspase-1 stimulation by NLRP3, a cleaved p20 fragment leads to the release of pro-inflammatory cytokines [9]. Furthermore, p20 also induces the cleavage of inactive gasdermin D (GSDMD) into the active GSDMD-NT fragment, which can bind to cell membranes in lung tissues to induce pyroptosis [10].
Triggering receptor expressed on myeloid cells-1 (TREM-1), a transmembrane immunoglobulin superfamily receptor, is a potent pro-inflammatory amplifier. It has been shown that TREM-1 mediates pyroptosis in the rat middle cerebral artery occlusion model [11]. Previous studies have shown that blocking TREM-1 activation can inhibit NLRP3 inflammasome activation, thereby reducing lipopolysaccharide (LPS)-induced ALI [12]. It has been reported that TREM-1 can contribute to neuroinflammatory injury in experimental subarachnoid hemorrhage via NLRP3 inflammasome-mediated pyroptosis [13]. However, the role of TREM-1 in TBI-ALI is not fully understood. Herein, we hypothesize that TREM-1 could activate NLRP3-related pyroptosis after TBI-ALI, and the inhibition of TREM-1 may be an effective treatment for TBI-ALI.
It is reported that ondansetron (OD), a 5-hydroxytryptamine type-3 (5-HT3R) receptor antagonist, reduces blood–brain barrier damage and brain edema to exert neuroprotective effects [14]. Studies have shown that OD reduces the release of 5-HT and exerts therapeutic effects in pain-related behaviors and neuroinflammation after TBI [15]. 5-HT3R exists on the surface of macrophages, and its antagonists have shown effects against NLRP3 activation and neuroinflammation in animal models of Alzheimer’s disease [16]. However, whether OD has a therapeutic effect on TBI-ALI and its underlying pharmacological mechanisms are unclear.
Therefore, using a lateral fluid percussion (LFP) brain injury rat model with pharmacological intervention, this study aims to evaluate the protective effect of ondansetron on TBI-ALI, and to explore whether the underlying mechanism is related to NLPR3 activation and pyroptosis regulated by TREM-1.
Material and methods
Animals
Adult male Sprague-Dawley rats (12 weeks old, 250–320 g) were obtained from the Laboratory Animal Center of Southern Medical University, P.R. China [Certification: SYXK (Guangdong) 2021-0041]. The rats were housed at a standard temperature of 24 ±1°C under a 12 h light-dark cycle (dark, 8:00 pm–8:00 am), with free access to food and water. Our animal experiments and operations were performed in accordance with the National Institutes of Health guidelines and were approved by the local Animal Care and Use Committee of Southern Medical University, Guangzhou, China.
Experimental groups and drug administration
According to different experimental purposes, rats were randomly assigned to different groups in accordance with the random number table. To clarify the effects of TBI on NLRP3 activation, pyroptosis, and TREM-1 expression in lung tissue, rats were divided into a sham group and a TBI group at different timepoints (24, 48, and 72 h following TBI). For TREM-1 intervention, rats were randomized to the sham group, TBI + NS (normal saline) group and TBI + LP17 (TREM-1 inhibitory peptide) group. To clarify whether OD has a protective effect following TBI-ALI, rats were randomized to the sham group, the TBI + NS group, and the TBI + OD group. LP17 (4 mg/kg, diluted in normal saline; Zhuantai Co. Ltd., Hangzhou, China) or OD (2 mg/kg; Oubei, Shandong, China) was, accordingly, applied intravenously 30 min after TBI with a pre-determined dose based on a previous study [17, 18]. For sample size of the animals, N = 3–5 per group, depending on the particular experiment.
Lateral fluid percussion brain injury operation
The rat model of TBI-ALI induced by LFP brain injury was constructed based on the model reported in our previous study but with a few modifications [7, 19]. In summary, rats were anesthetized with isoflurane and placed in a stereotaxic frame. Their scalp and temporal muscles were removed. The skull was then pierced through at 2.5 mm lateral to the sagittal sinus and centered between the bregma and lambda to create a 4.8 mm cranial cavity. A hollow female Luer-Lok fitting was inserted directly into the dura region and secured using dental cement. The female Luer-Lok was connected to the fluid percussion injury device via a transducer (Biomedical Engineering Facility, Medical College of Virginia, USA). A metal pendulum was released from a pre-determined height, resulting in a swift injection of normal saline into the closed cranial cavity, leading to TBI. An oscilloscope was used to elicit, control, and record a pulse of increased intracranial pressure lasting 21–23 ms (Agilent 54622D, MEGAZoom, Germany). By fluctuating the exertion generated by the pendulum, the severity of the injury was altered. This experiment leads to severe damage of 3.0 ±0.2 atm [20]. Sham rats were exposed to identical surgical procedures, including craniotomy, but not injured. Brain tissue samples from the injury site and lung tissue samples of rats were collected for future experiments.
Histopathological examination
Rat lung tissues were fixed in 4% paraformaldehyde for 48 h before paraffin embedding. Sections were sliced and stained with hematoxylin and eosin (H&E), and examined under a light microscope (Olympus, Tokyo, Japan) to observe lung injury. A standardized and validated semiquantitative scoring system in accordance with the American Thoracic Society workshop report was applied to evaluate lung injury as previously published and as described in Table I [21, 22].
Table I
Lung injury scoring system [22]
Wet/dry ratio estimation
The lungs were weighed immediately for wet weight after euthanizing the rats. The dry weight of lung tissues was determined after drying at 60°C for 24 h. Lung wet/dry ratios indicate pulmonary edema and congestion.
Western blotting
Protein was isolated from lung tissues, separated by 8–12% SDS-PAGE under reducing conditions before being transferred to 0.45 μm polyvinylidene difluoride membranes. The membranes were incubated with the following primary antibodies: TREM-1 (1 : 1000; DF6091, Affinity Bioscience, Jiangsu, China), Interleukin 6 (IL-6) (1 : 1000; DF6087, Affinity Bioscience, Jiangsu, China), tumor necrosis factor-a (TNF-α) (1 : 1000; AF7014, Affinity Bioscience, Jiangsu, China), HMGB1 (1 : 5000; orb195321, Biorbyt, San Francisco, CA, USA), IL-18 (1 : 1000; DF6562, Affinity Bioscience, Jiangsu, China), IL-1β (1 : 1000; YT2322, Immunoway, Jiangsu, China), ASC (1 : 1000; DF6304, Affinity Bioscience, Jiangsu, China), NLRP3 (1 : 1000; #15101, Cell Signaling Technology, MA, USA), GSDMD (1 : 500; sc-393656, Santa Cruz Biotechnology, CA, USA), and caspase-1 (1 : 500; sc-56036, Santa Cruz Biotechnology, CA, USA) at 4°C overnight. Subsequently, the membranes were washed with TBST (Tris-buffered saline with Tween) three times and incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG antibody as the secondary antibody (1 : 5000; CW0102S, Jiangsu, China). Enhanced chemiluminescence was used to detect bound antibodies (Meilunbio, Dalian, Liaoning, China) using the Tanon imaging system (Tanon-5200, Shanghai, China). The internal control was β-actin gene expression.
Immunofluorescence
After fixation and dehydration, the frozen sections of rat lung tissues were prepared. Primary alveolar macrophages (AMs) from bronchoalveolar lavage fluid (BALF) were seeded at 3 × 104 cells per well on 24-well glass slides and incubated for 6 h to enable attachment. The tissue sections and cells were fixed with 4% methanol, perforation with 0.1% Triton x-100 and blocked for 1 h in 5% BSA and 0.1% Tween-20 in PBS at room temperature. The slides were incubated with primary antibodies against GSDMD (1 : 100; AF4012, Affinity Bioscience, Jiangsu, China) and CD68 (1 : 500; ab283654, Abcam, Cambridge, UK) at 4°C overnight. AMs were incubated with primary antibodies against CD68 and TREM-1 (1 : 100). Subsequently, second antibodies conjugated to Alexa Fluor-647 (1 : 200; ab150075, Abcam, Cambridge, UK) or fluorescein isothiocyanate (1 : 300; B40593, Thermo Fisher Scientific, CA, USA) were applied. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI; 1 : 500; Beyotime Institute of Biotechnology, Shanghai, China). The images were obtained by laser scanning confocal microscope (Zeiss LSM780, Thuringia, Germany).
Real-time PCR
Total RNA was extracted from lung tissues using the RNAiso Plus reagent (Takara, Otsu, Japan). First-strand cDNA was synthesized using the reverse transcription kit (Vazyme Biotech, Nanjing, China), according to the manufacturer’s protocols. In the presence of fluorescent dye (SYBR Green I), the real-time PCR reaction was performed using quantitative PCR (Applied Biosystems, Thermo Fisher Scientific, CA, USA). TREM-1 relative mRNA expression was normalized using β-actin and studied using the 2-ΔΔCt method. The primer sequences (Tsingke Biotechnology, Beijing, Jiangsu, China) are listed below (F – forward primer, R – reverse primer):
Immunohistochemical analysis
Formaldehyde-fixed lung tissues were embedded in paraffin and cut into 4 μm thick specimens before being deparaffinized with xylene and incubated with 3% H2O2 for 10 min. Antigens were salvaged by microwaving the specimens in citric acid buffer. Lung sections were incubated overnight at 4°C with a rabbit anti-TREM-1 polyclonal antibody (1 : 100, Affinity Bioscience, Jiangsu, China). After being rinsed , the sections were incubated with HRP-conjugated secondary antibody (#ab205718, Abcam, Cambridge, UK). Tissue sections were then stained with 3,3’-diaminobenzidine (DAB) and counterstained with hematoxylin. Images were captured using a light microscope (Olympus, Tokyo, Japan).
BALF protein
BALF was harvested by washing the lung tissues three times with 5 ml of phosphate-buffered saline (PBS) using a tracheal cannula and centrifuging them at 2000 rpm for 10 min at 4°C. The protein concentration in the cell-free supernatants was determined using a BCA protein assay kit (Beyotime Institute of Biotechnology, Shanghai, China).
Statistical analysis
SPSS 26.0 statistical software (IBM, Armonk, NY, USA) was used for all statistical analyses. One-way ANOVA was used to determine the statistical differences between groups, and the Student-Newman-Keuls test was utilized for post-hoc multiple comparisons. The results are presented as the mean ± standard deviation. Statistical significance was defined as p < 0.05.
Results
TBI-activated NLRP3 inflammasome and pyroptosis in lung tissues
We confirmed whether TBI could induce ALI in the rat model. As predicted, on comparison with the sham group, a high percentage of apoptotic cells was observed in injured brain tissues composed of the cortex and hippocampus of TBI rats (Figure 1 A). Moreover, interstitial edema, severe alveolar hemorrhage, and extensive inflammatory cell infiltration were observed in rat lungs at different time points (24 h, 48 h, and 72 h), accompanied by gradually increasing lung injury scores (Figures 1 B, C) and wet/dry ratios (Figure 1 D).
Figure 1
TBI induced NLRP3 inflammasome and pyroptosis in lung tissue. A – Toluidine blue staining of the cortex and hippocampus. Scale bar = 250 μm for the upper row and the zoom of the outlined areas for the lower row, arrows for apoptotic neurons, N = 4. B – H&E staining of lung tissues at 24 h, 48 h, and 72 h post-TBI. scale bar = 100 μm for original magnification. Green arrows for interstitial edema; blue arrows for inflammatory infiltration; black arrows for hemorrhage. N = 4. C – Lung injury scores based on the histopathological staining of lungs. D – Wet/dry weight ratio of the lungs. N = 4. E – Representative western blots of NLRP3, Cleaved caspase-1, ASC, IL-18, and IL-1β in lung tissues. Densitometric analysis of (F) NLRP3, (G) Cleaved caspase-1, (H) ASC, (I) IL-18, and (J) IL-1β. N = 3. K – Representative western blots of full-length GSDMD (GSDMD-FL) and GSDMD-N domain (GSDMD-NT) protein levels in lung tissues. L – Densitometric analysis of GSDMD-NT. N = 3. M - Immunofluorescence of GSDMD expression in lung tissue after TBI. Scale bar = 20 μm. Results were normalized to β-actin. The values are presented as mean ± standard deviation (SD). *P < 0.05, **P < 0.01, ***P < 0.001 vs. Sham group
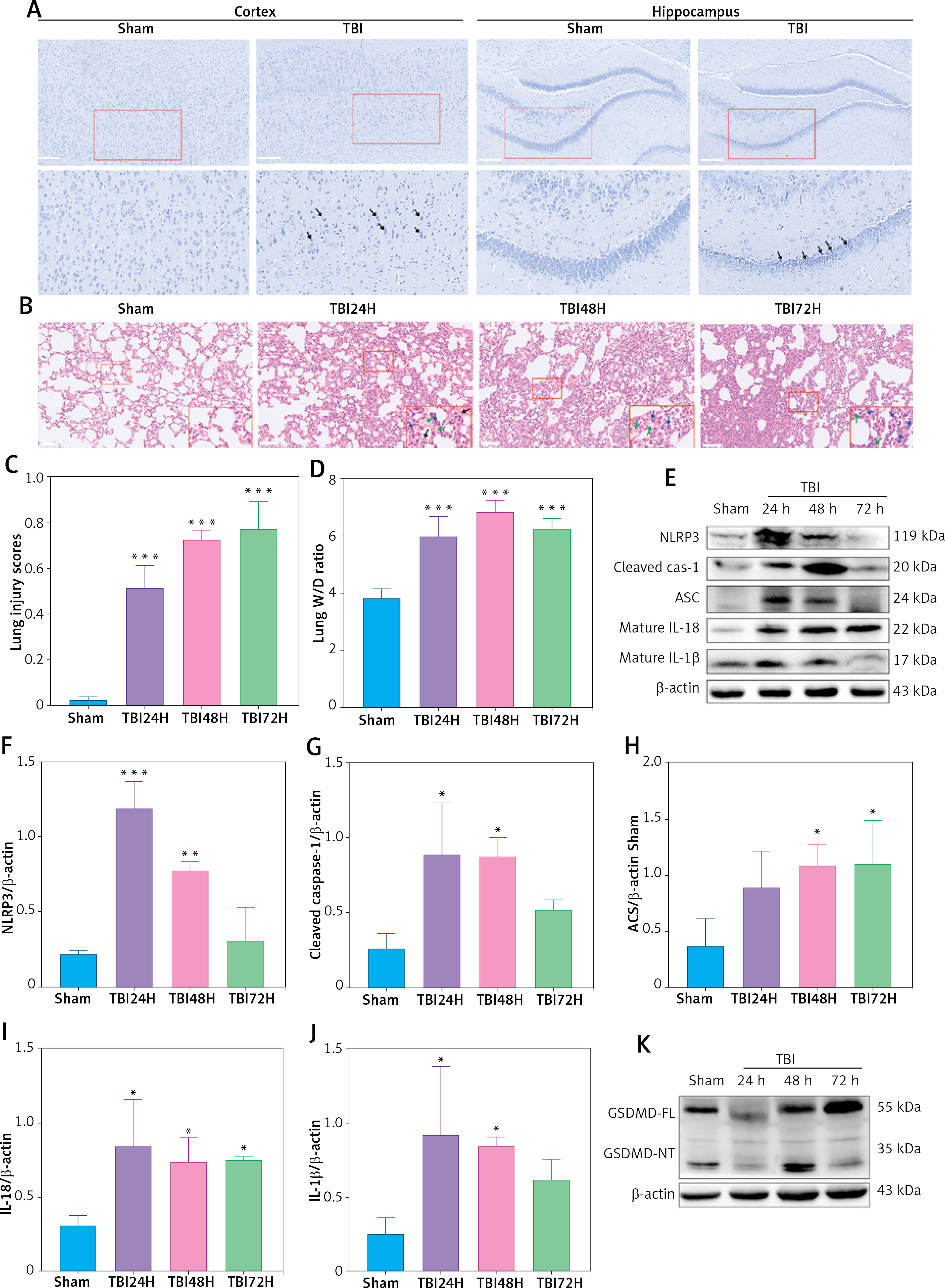
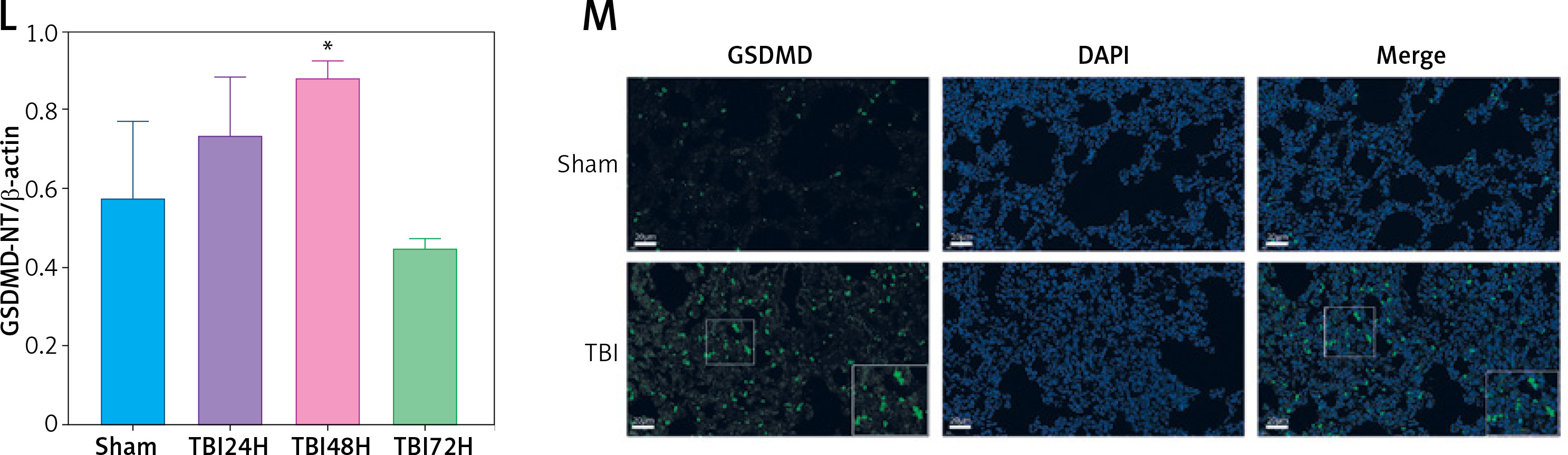
We next explored the effect of TBI on the NLRP3 inflammasome and pyroptosis in the lung tissue. The expression of NLRP3 inflammasome-related proteins, including NLRP3, apoptosis-associated speck-like protein (ASC), and cleaved caspase-1, as well as the contents of mature IL-1β and mature IL-18, considerably increased in lung tissue of rats within 72 h after TBI (Figure 1 E). Notably, the levels of full-length GSDMD and N-terminal GSDMD (GSDMD-NT, an active form of GSDMD) increased significantly at 48 h after TBI (Figure 1 K) with markedly elevated GSDMD expression in immunofluorescence analysis (Figure 1 M).
TREM-1 inhibition attenuated TBI-ALI
To explore the role of TREM-1 in TBI-ALI, we determined the mRNA and protein levels of TREM-1 in lung tissue after TBI. The expression of TREM-1 mRNA (Figure 2 A) and protein levels (Figures 2 B, C) were significantly upregulated following TBI in rats compared to the sham group. Immunohistochemistry results confirmed increased TREM-1 expression at 48 h after TBI (Figure 2 D). Correspondingly, the expression of pro-inflammation cytokines increased significantly after TBI (Figures 2 E–H).
Figure 2
Inhibition of TREM-1 attenuated TBI-ALI. A – mRNA expression of TREM-1 in lung tissue at 24, 48, and 72 h post-TBI. N = 3. B – Representative western blots of TREM-1 in lung tissue at 24, 48, and 72 h post-TBI. C – Densitometric analysis of TREM-1. N = 3. D – Immunohistochemical staining of TREM-1 at 48 h post-TBI. Scale bar = 50 μm, N = 4. E – Representative western blots of pro-inflammatory cytokines IL-6, TNF-α, and HMGB1 in lung tissue at 24, 48, and 72 h post-TBI. Densitometric analysis of (F) IL-6, (G) TNF-α, and (H) HMGB1. N = 3. I – Representative images of H&E-stained lung tissue from the sham group, TBI + NS group, and TBI + LP17 group are shown. Scale bar = 100 μm, N = 4. J – Lung injury scores based on histopathological staining of lungs. K – The wet/dry ratio of the lungs. N = 4. L – Total protein in the BALF. N = 4. M – Representative western blots of TREM-1 and NLRP3 inflammasome components. Densitometric analysis of (N) TREM-1, (O) NLRP3, (P) Cleaved caspase-1, (Q) ASC, (R) IL-1β, (S) IL-18. Results normalized to β-actin. N = 4–5. The values are presented as mean ± standard deviation (SD). *P < 0.05, **p < 0.01, ***p < 0.001 vs. Sham group. #P < 0.05, ##p < 0.01 vs. TBI + NS group
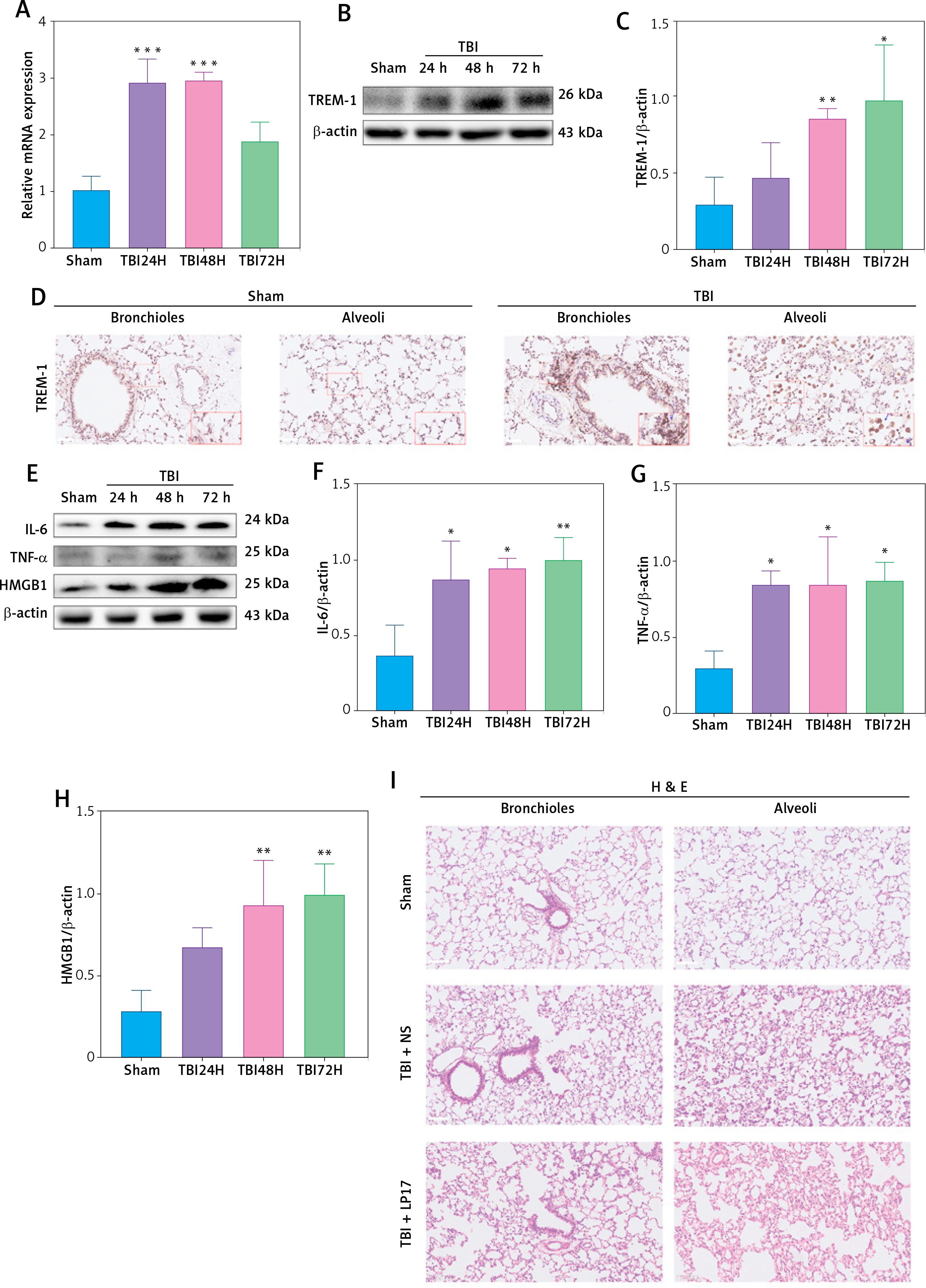
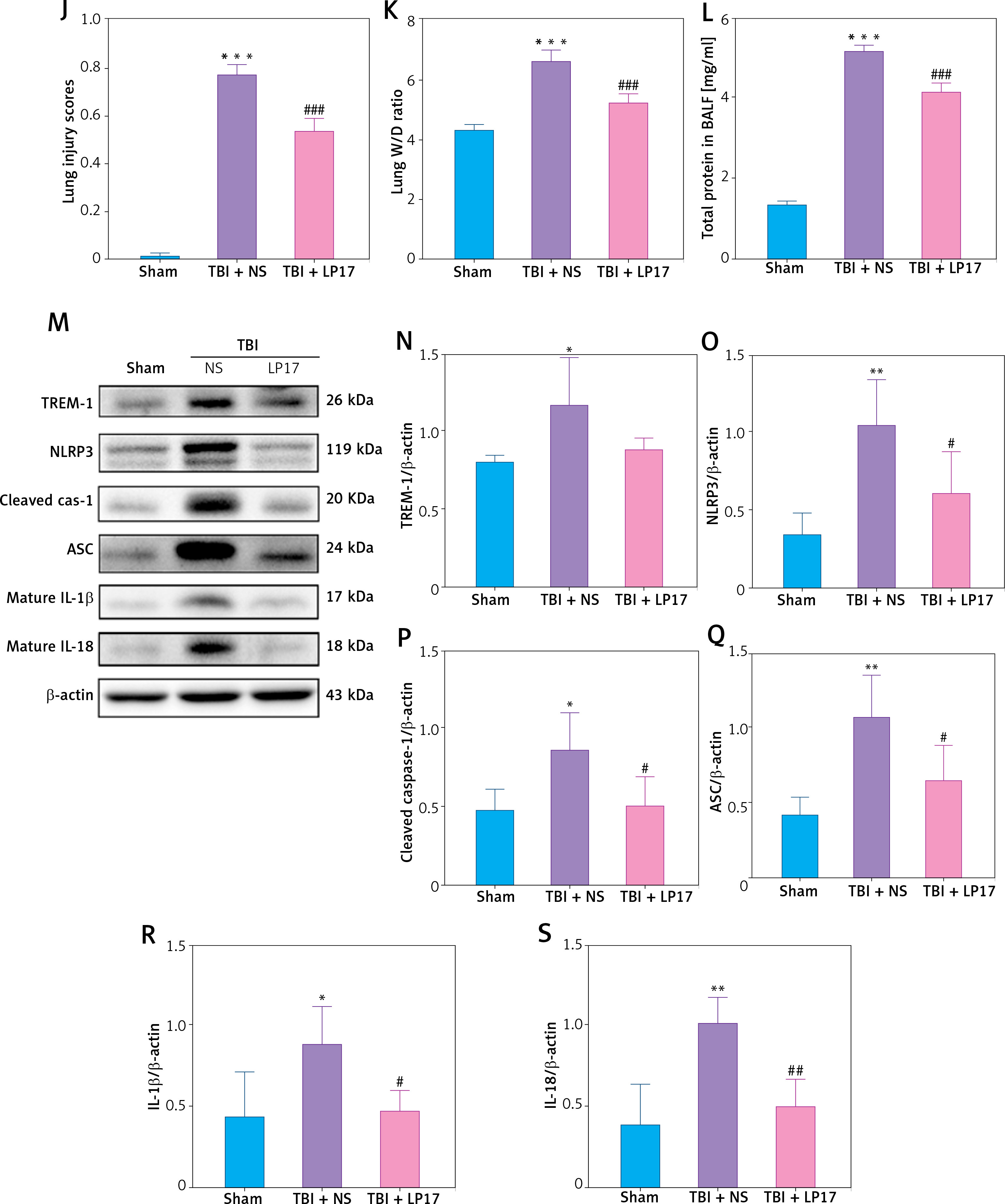
To further clarify the role of TREM-1 in TBI-ALI, we administered LP17, a chemical inhibitor of TREM-1, to TBI rats. LP17 reduced alveolar wall thickening and immune cell infiltration (Figure 2 I) and lowered lung injury scores (Figure 2 J), lung wet/dry ratios (Figure 2 K), and BALF total protein content (Figure 2 L) after TBI. LP17 also inhibited the expression of NLRP3, ASC, cleaved caspase-1, mature IL-1β, and mature IL-18 (Figures 2 M–S). These results collectively suggest that the increased NLRP3 inflammasome expression after TBI-ALI is dependent on TREM-1 activation.
OD suppressed NLRP3 inflammasome and pyroptosis and ameliorated ALI
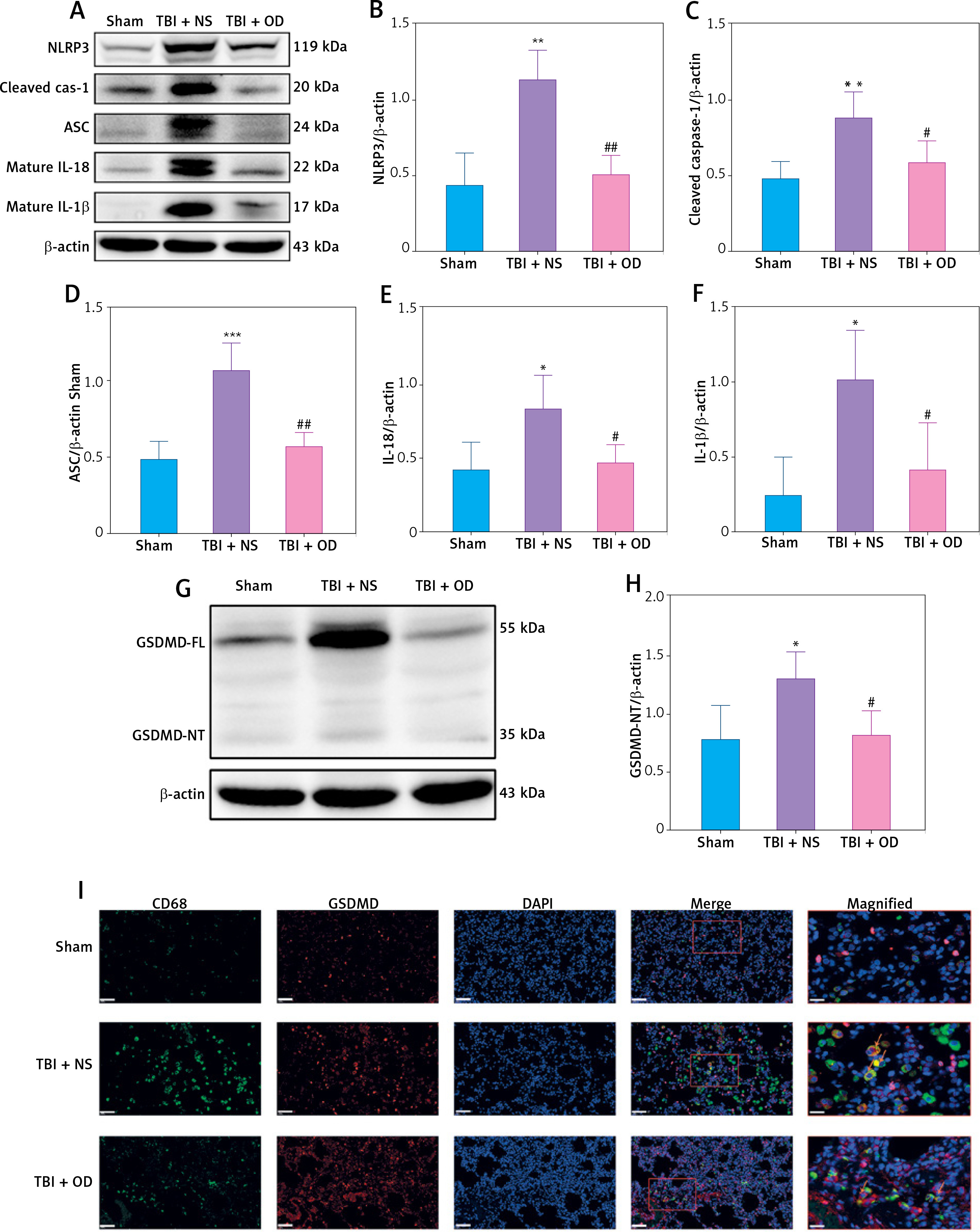
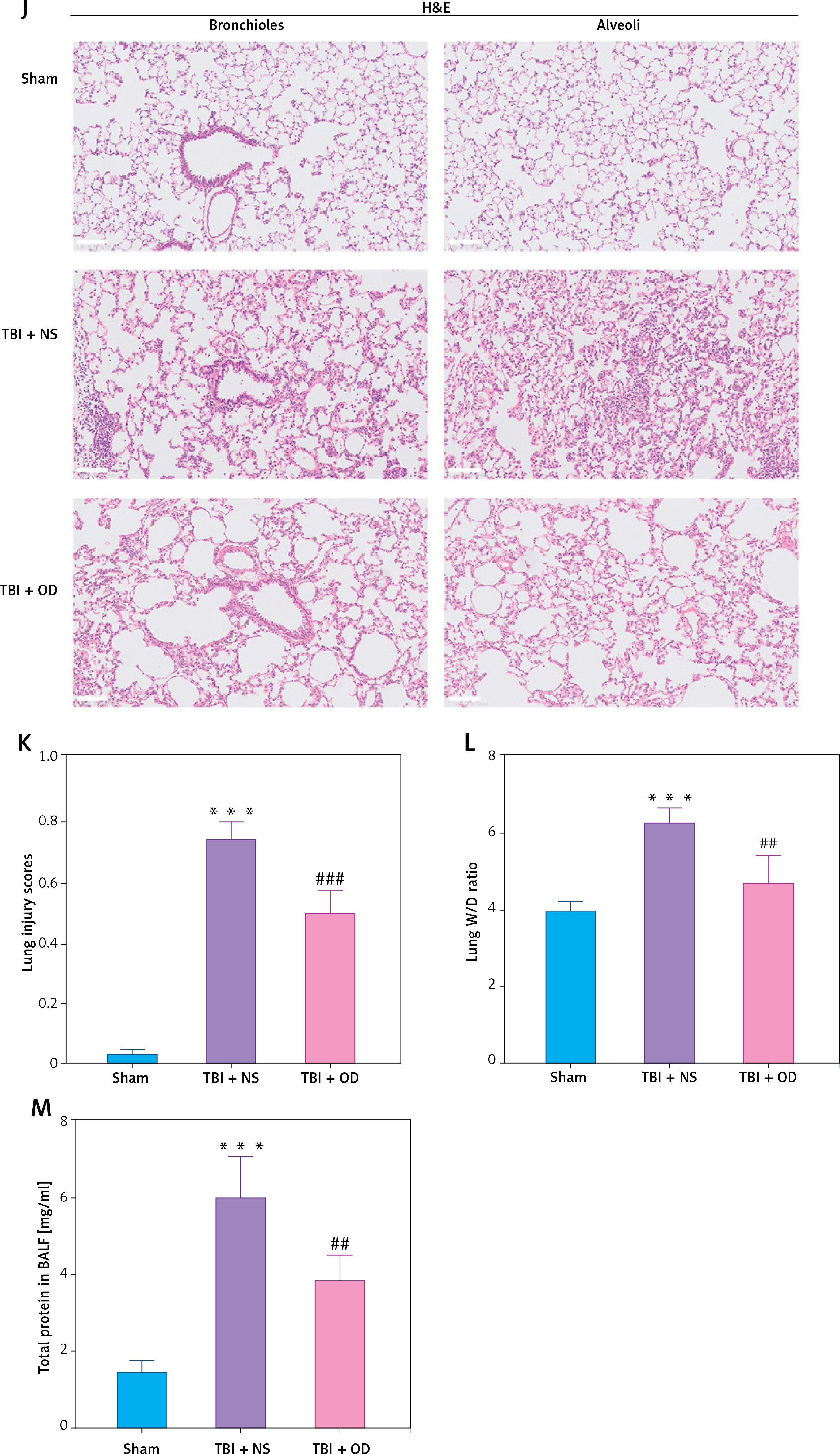
To confirm the therapeutic effect and mechanism of OD on TBI-ALI, we tested NLRP3 inflammasome expression in lung tissue. Compared with that in the TBI + NS group, OD treatment markedly reduced the protein levels of NLRP3, ASC, caspase-1, IL-1β, and IL-18 (Figures 3 A–F) in the TBI+OD group. Moreover, OD considerably lowered the protein levels of GSDMD-FL and GSDMD-NT (Figures 3 G, H). Consistent results were found in the immunofluorescence assay. As pyroptosis of macrophages is essential for the pathogenesis of ALI, we focused on macrophages and observed an increase in the number of CD68-positive macrophages in lung tissue from TBI rats (Figure 3 I). OD reduced GSDMD expression in CD68 positive macrophages (Figure 3 I). In addition, OD treatment reduced immune cell infiltration, alveolar wall thickening, lung injury scores (Figures 3 J, K), lung wet/dry ratios, and BALF protein contents (Figures 3 L, M).
Figure 3
Ondansetron reduces TBI-ALI in rats. A – Representative western blots of NLRP3 components. Densitometric analysis of (B) NLRP3, (C) Cleaved caspase-1, (D) ASC, (E) IL-18, and (F) IL-1β. Results normalized to β-actin, N = 4. G – Representative western blots of GSDMD protein expression. H – Densitometric analysis of GSDMD-NT. Results normalized to β-actin, N = 4. I – Representative images of lung tissue after immunostaining for CD68 and GSDMD. Scale bar = 50 μm, N = 4. K – Lung injury scores following OD treatment. L – The wet/dry ratio of the lungs. N = 4. M – Total protein in the BALF. N = 4. The values presented as mean ± standard deviation (SD). *P < 0.05, **p < 0.01, ***p < 0.001 vs. sham group. #P < 0.05, ##p < 0.01 vs. TBI + NS group
OD inhibited TREM-1 expression in AMs
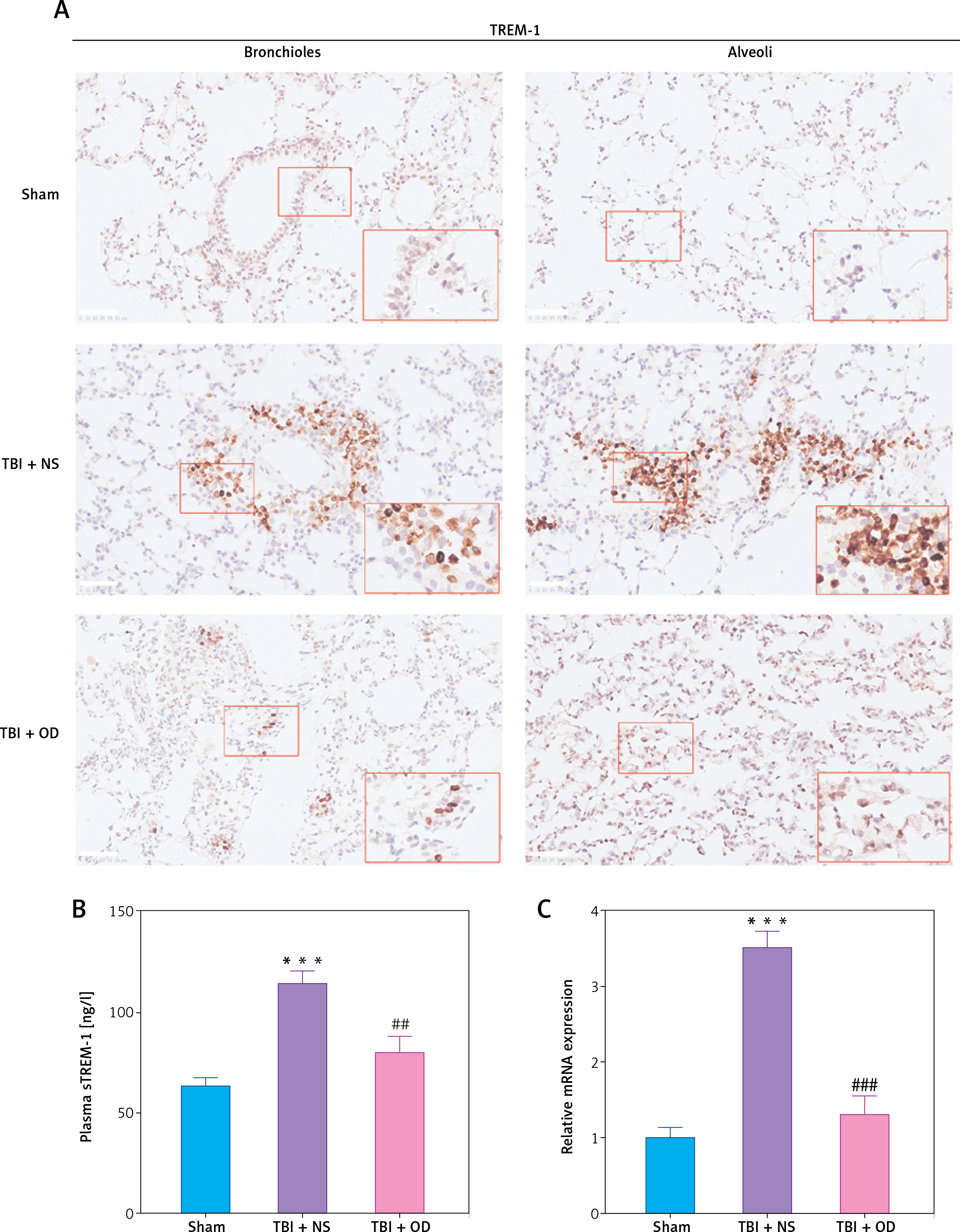
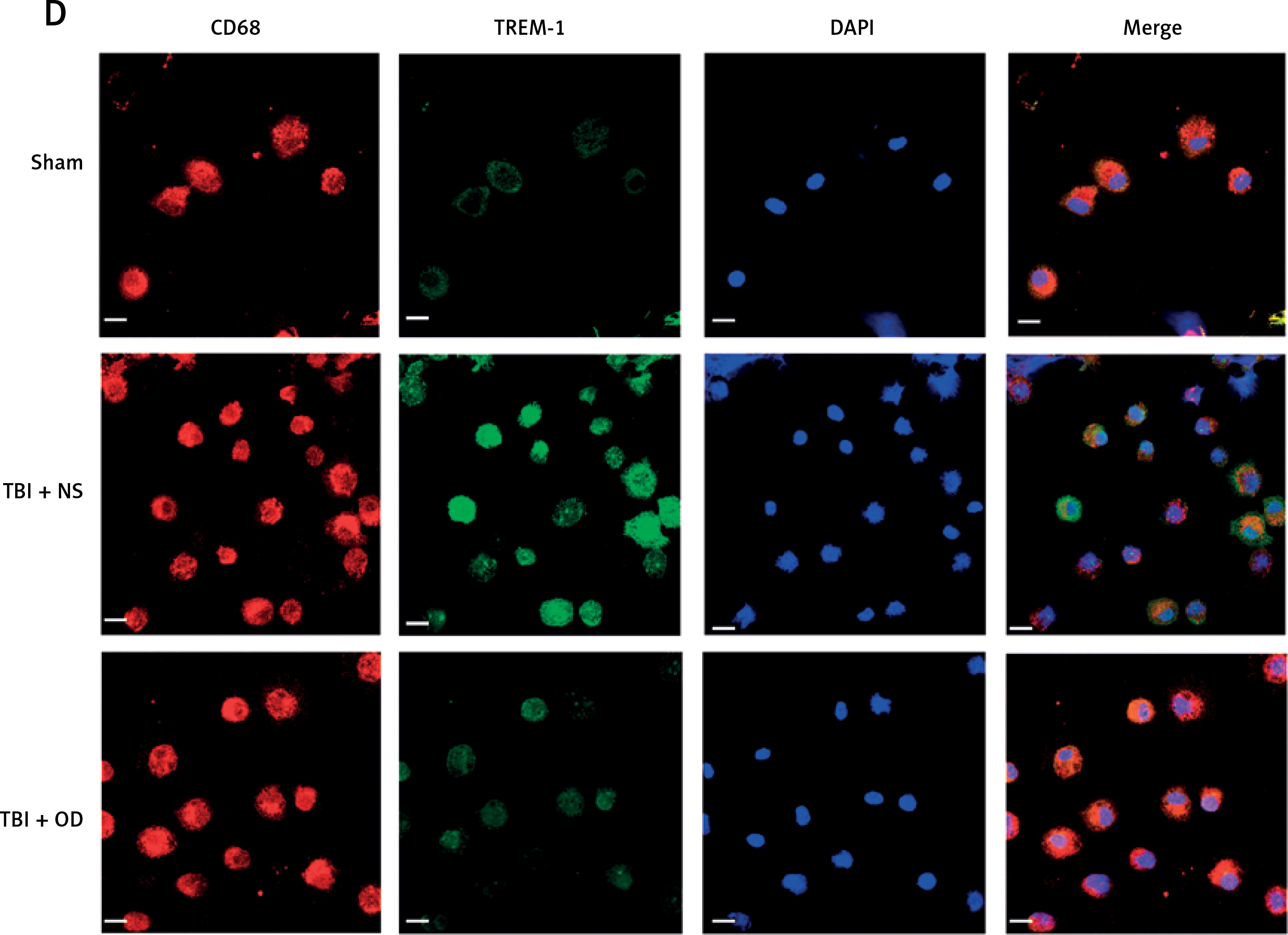
Next, we explored the effect of OD on TREM-1 expression in AMs. As expected, OD inhibited TREM-1 expression, as observed in immunohistochemical results (Figure 4 A), lowered plasma sTREM-1 levels (Figure 4 B), and decreased TREM-1 mRNA expression in AMs (Figure 4 C). Immunofluorescent cells in bronchial fluid consistently exhibited decreased TREM-1 expression in AMs upon OD treatment (Figure 4 D).
Figure 4
Ondansetron inhibits TREM-1 expression in alveolar macrophages. A – TREM-1 staining of bronchioles and alveoli in histology sections. Scale bar = 50 μm, N = 4. B – sTREM-1 plasma levels in sham group, TBI + NS group, and TBI + OD group. N = 4. C – TREM-1 mRNA expression after OD administration. N = 4. ***P < 0.001 vs. sham group. ###P < 0.001 vs. TBI + NS group. D – Representative images of isolated AMs in BALF after immunostaining for CD68 and TREM-1. Scale bar = 50 μm. N = 3.The values are presented as mean ± standard deviation. ***P < 0.001 vs. sham group. ###P < 0.001 vs. TBI + NS group
Discussion
Our study showed that OD can help protect against TBI-ALI by inhibiting NLRP3 inflammasomes/pyroptosis in macrophages. Regarding the mechanism, we discovered that TREM-1 protein expression increases after TBI and that TREM-1 activates NLRP3 inflammasome/pyroptosis in lung tissues, thereby worsening TBI-ALI. Furthermore, the protective effect of OD on TBI-ALI may be related to the inhibition of TREM-1 expression.
Excessively released inflammatory factors are crucial in development of TBI-ALI [23]. A study by Kerr et al. proposed the concept of the neuro-respiratory-inflammasome axis, emphasizing the role of inflammasome signaling in TBI-ALI [24]. Our study also supports the existence of various activated inflammasome-related cytokines such as IL-6, IL-1β, IL-18, TNF-α, and HMGB1 in lung tissue after TBI. Contrary to what Kerr et al. reported, we found that the time to ALI after TBI was 48 h, much later than the 4 h reported by them [8]. Another study found that the lung injury induced by the midline hydraulic impact injury model in CD-1 mice could last from 1 h to the seventh day after TBI [25], which means that different strains of animals and TBI modeling methods may have triggered various progress changes in TBI-ALI.
TREM-1 is involved in the inflammatory response process of various diseases. TREM-1 inhibition has shown effective therapeutic effects in patients experiencing septic shock in a clinical phase two trial [26]. In septic mice, highly expressed TREM-1 caused severe lung tissue damage and upregulated the levels of pro-inflammatory factors [27]. Our study showed that increased TREM-1 protein expression is involved in TBI-ALI, supporting the use of TREM-1 as a biomarker for predicting ALI. In addition to amplifying inflammation in a TLR-dependent manner, TREM-1 also accelerated pyroptosis by activating the inflammasome. Studies have confirmed that in a rat model of subarachnoid hemorrhage, TREM-1 could induce microglial pyroptosis via upregulation of the NLRP3 inflammasome and promoting neuroinflammation [13, 28]. In Kerr’s study, TBI induced expression of inflammatory factors and activated inflammasome-induced pyroptosis, which primarily concentrated in type II alveolar epithelial cells [29]. Our research findings indicate that TBI results in pyroptosis in different cell types found in lung tissue. Interestingly, in macrophages specifically, the process of pyroptosis is associated with an increase in the expression of TREM-1. However, after the administration of a TREM-1 inhibitor, lung tissue pathological damage and pyroptosis were significantly alleviated. These observations suggest that TREM-1 could play a role in the pathogenesis of TBI-ALI and highlight the therapeutic potential of TREM-1 inhibition in TBI-ALI.
Many studies have shown that 5-HT3R inhibitors possess anti-inflammatory and immunomodulatory properties [30]. Research has suggested that the inhibition of 5-HT3R can lead to a reduction in macrophage-mediated inflammatory responses in cases of sepsis [31]. In sepsis-induced neuroinflammation, 5-HT3R inhibitor reduces the release of pro-inflammatory cytokines in microglia [32]. In addition, 5-HT3R inhibitor has been reported to alleviate the release of pro-inflammatory cytokines via the NLRP3 inflammasome to attenuate sepsis-induced ALI [33]. A study reported that OD, a 5-HT3R inhibitor commonly used in clinical practice, can modulate descending pain after TBI [15]. Our experimental results showed that OD alleviated TBI-ALI by inhibiting NLRP3 activation and pyroptosis. Additionally, OD significantly inhibited TREM-1 expression in macrophages and lung tissue, suggesting a potential role for TREM-1 in the effect of OD on TBI-ALI.
Our study has some limitations. First, the mechanism of the specific regulatory effect of TREM-1 on NLRP3-related pyroptosis needs further study. Second, although our results suggest that TREM-1 contributes to macrophage pyroptosis in lung tissue after TBI, further investigation is needed to determine the expression and function of TREM-1 in other cell types involved in this process. Finally, our research suggests that OD may protect against TBI-ALI by inhibiting TREM-1 expression in macrophages. However, further investigation is needed to determine the exact molecular mechanisms underlying this effect and to determine whether other cell types are involved. Regardless, our study confirmed that OD could alleviate TBI-ALI. As regards the mechanism, TREM-1 promotes TBI-ALI via the NLRP3-related pyroptosis pathway, and the protective effect of OD on TBI-ALI may be related to the inhibition of TREM-1-mediated pyroptosis. Ondansetron and TREM-1 inhibitors may be promising therapeutic agents for the treatment of TBI-ALI. While further research is needed to confirm the efficacy and safety of these treatments in humans, the results of these studies provide a promising avenue for the development of effective therapies for TBI-ALI.