Introduction
Lipoprotein(a) [Lp(a)] was discovered in 1963 by Norwegian scientist Kåre Berg [1]. Over the following 60 years, knowledge about lipids and cardiovascular diseases (CVD) progressed significantly. Over the past decade, it has become increasingly clear that Lp(a) is a key component of residual cardiovascular disease risk [2]. Lp(a) has been shown to be strongly and independently associated with atherosclerotic cardiovascular disease (ASCVD) and the occurrence of aortic valve stenosis (AVS). Lp(a) is also associated with peripheral artery disease (PAD) and, to a lesser extent, with ischaemic stroke (IS) and heart failure (HF) [3]. Recent studies also suggest that higher levels of Lp(a) may be associated with an increased risk of atrial fibrillation (AF), but this requires further confirmation in studies [4–6]. In contrast, the association between Lp(a) and the risk of venous thromboembolism is not supported by most recent studies [7, 8].
Elevated Lp(a) levels ≥ 50 mg/dl (≥ 125 nmol/l) are estimated to occur in more than 1.5 billion people worldwide [9]. However, tests on this parameter are performed far too rarely [10–12], most often due to insufficient knowledge. There are currently no dedicated medicines that target the reduction of Lp(a) concentrations and available lipid-lowering treatment can only modify the concentration of this parameter to a certain extent (20–30%). The aim of this Expert Opinion prepared by the two most important scientific societies in Poland in the relevant area is to provide clinical recommendations for the determination and treatment of elevated Lp(a) levels.
Lipoprotein(a) molecular structure
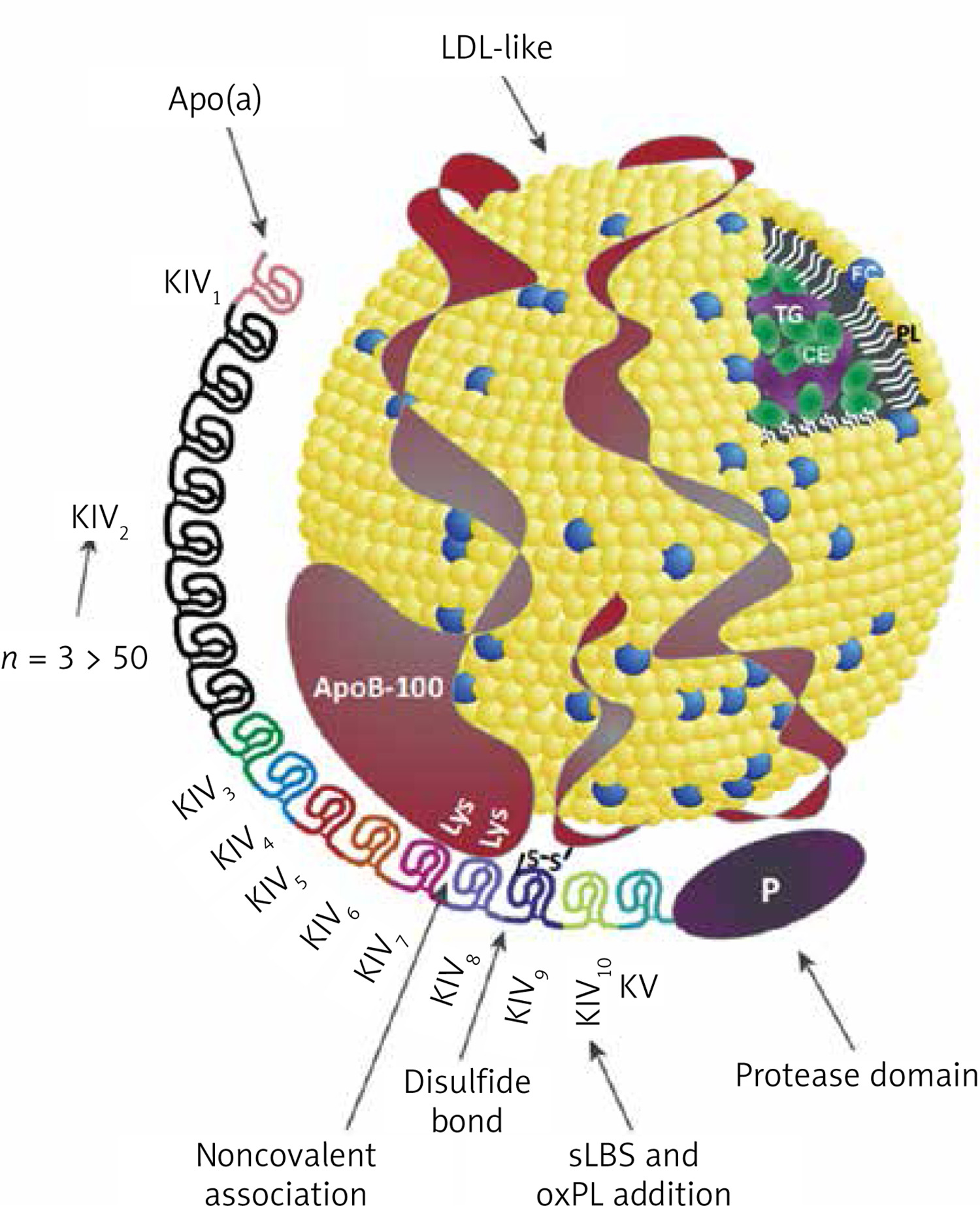
Lp(a) is a lipoprotein composed of a low-density lipoprotein (LDL)-like particle and a specific apolipoprotein(a) [apo(a)] [13] (Figure 1). Apo(a) is synthesised in hepatocytes, a characteristic feature of apo(a) is the presence of protein triple-loop (kringle) structures. Apo(a) is composed of a single kringle V (KV) domain, a non-reactive serine protease-like domain, and 10 KIV subtypes (KIV1-KIV10), among them the KIV2 subtype, which occurs in < 3 to > 50 copies [14, 15] (Figure 1). The number of KIV2 repeats is determined by the LPA gene [16]. Apo(a) is covalently linked to apoB100 via a single disulphide bond involving a unique unpaired cysteine residue (Cys4057) in the KIV9 domain [17], and via non-covalent interactions involving the KIV7 and KIV8 domains [18]. The KIV10 domain contains a strong lysine binding site (sLBS) that mediates interactions between apo(a) and biological ligands such as fibrin [19]. Importantly, KIV10 also contains a site to which oxidised phospholipids (OxPL) are covalently attached [20] (Figure 1). Most individuals (~80%) have two apo(a) alleles of different magnitudes, each inherited from one parent [21]. Thus, a person may have two types of molecules: two large, two small or different sized apo(a) molecules [22].
Figure 1
Structure of the lipoprotein(a) molecule, reprinted and modified from [22] with permission (Licence number: 5705320305220)
P – protease-like domain, PL – phospholipids, FC – free cholesterol, CE – cholesterol esters, TG – triglycerides.
Lp(a) is rich in cholesterol esters and phospholipids and contains lower amounts of free cholesterol and triglycerides [23]. Lp(a) particles are the main carrier of OxPL among apolipoprotein B (apoB)-containing lipoproteins [24]. Lp(a) is predominantly removed and catabolised by the liver (and to a lesser extent by the kidney) via a number of putative pathways mediated by LDL receptors (LDLR), class B1 scavenger receptors (SRB1) and the plasminogen receptors PLGR and PLGRKT [25, 26].
The physiological function of Lp(a) is still not known [16]. The mechanism of pathophysiological action of Lp(a) associated with CVD risk involves multiple pathways, most notably an apoB-mediated proatherogenic effect [27, 28] and a pro-inflammatory effect through its oxidised phospholipid content [29–31]. The chemoattractant for type 1 monocytes (MCP-1) present on the Lp(a) molecule increases the migration of monocytes across the endothelial cell barrier into the vessel wall. At the same time, Lp(a) stimulates monocytes to increase production and release of pro-inflammatory cytokines (including interleukin (IL)-1β, IL-6, IL-8, tumour necrosis factor-α [TNF-α]) [30]. Lp(a) is also associated with AVS/CVD risk through the proinflammatory and procalcifying effects of OxPL, which are thought to be able to enter the aortic valve through binding by apo(a). Lipoprotein-associated phospholipase A2 (Lp-PLA2) and autotaxin, enzymes present on Lp(a), are also likely to be involved in the pathogenesis of AVS/CVD [32]. It is worth noting here that the Lp(a) molecule has a high homology (75–99%) with plasminogen, but lacks protease activity, and was therefore assumed to inhibit fibrinolysis and show prothrombotic potential [33], but this has not been confirmed by recent studies on the effect of lowering Lp(a) on fibrinolytic activity or by studies on the effect of elevated Lp(a) levels on venous thrombosis [7, 8].
Lipoprotein(a) and cardiovascular disease
Results from a number of prospective, epidemiological, genetic, and randomised Mendelian randomization (MR) studies have shown that elevated Lp(a) levels are independently and causally associated with ASCVD and aortic valve stenosis (AVS) [34–42]. Results from recent studies show that exposure to higher concentrations of Lp(a) has the greatest effect on the risk of myocardial infarction (MI) and AVS, while the effect on the risk of ischaemic stroke, PAD, HF, CVD mortality and overall mortality is significant but somewhat weaker (Figure 2) [3, 43, 44].
Figure 2
Association between high plasma lipoprotein(a) levels and CVD (risk increase based on analysis of genetically determined increase in Lp(a) risk by one standard deviation according to: [63])
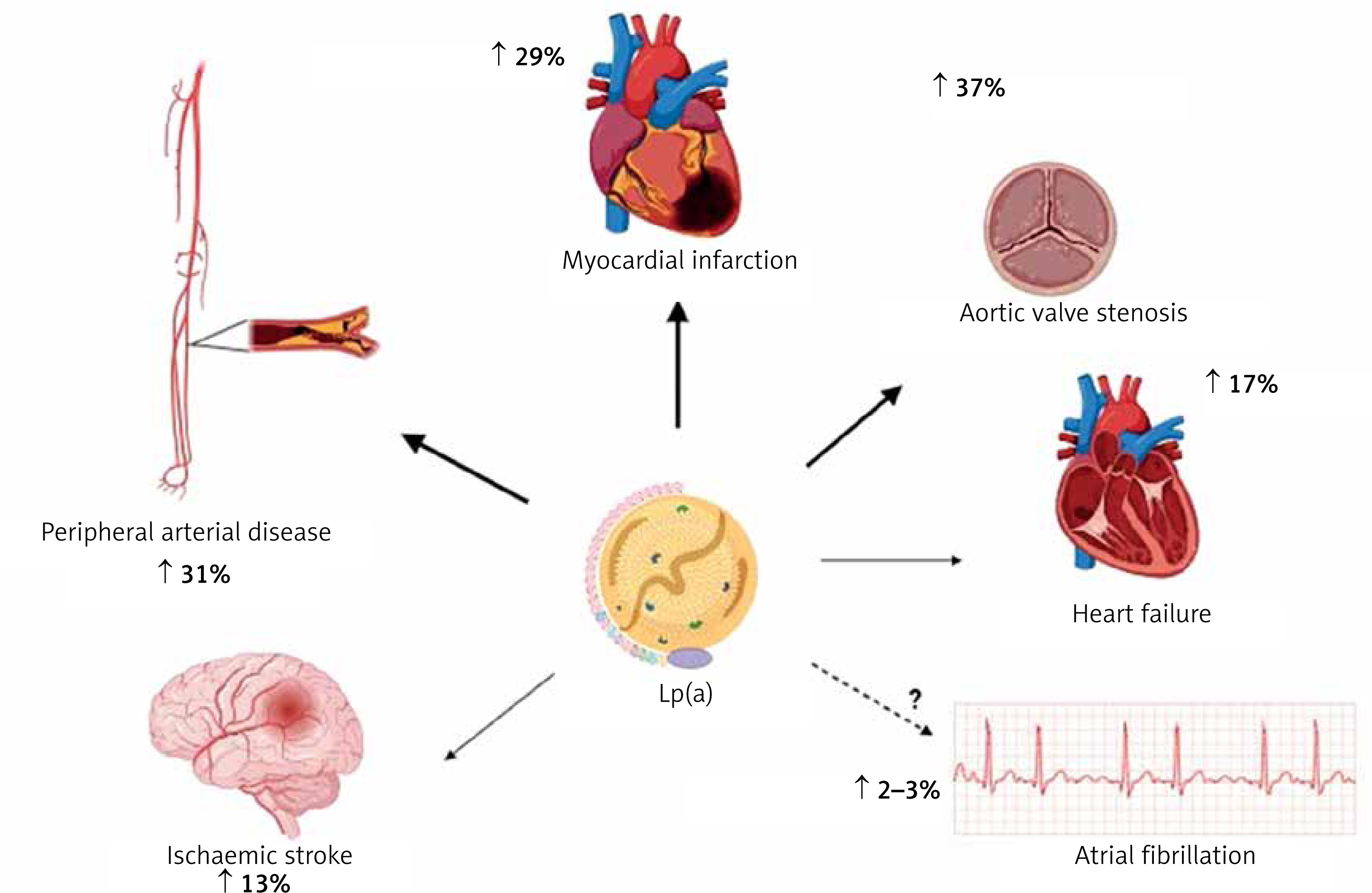
An analysis of UK Biobank data covering 460,000 people showed that the risk of ASCVD was 11% higher for each Lp(a) increment of 50 nmol/l (−20 mg/dl; risk ratio 1.11 per 50 nmol/l [95% CI: 1.10–1.12]) [45]. In individuals with very high Lp(a) levels (e.g. > 180 mg/dl [(450 nmol/l)]), the risk of ASCVD may be comparable to those with a family history of premature CVD or those with heterozygous familial hypercholesterolaemia (heFH) [46]. In a study on a large population of ~50,000 people, high Lp(a) levels significantly correlated with an increased incidence of ischaemic stroke [47]. A study in a Danish population of 69,764 participants showed that Lp(a) levels > 69 mg/dl (−172 nmol/l) were associated with a higher risk of death from CVD causes, and Lp(a) levels > 93 mg/dl (−232 nmol/l) resulted in a higher risk of death from any cause [47]. The association of Lp(a) with the risk of overall and CVD-related mortality has also been confirmed by other studies [48]. A recent study showed that each 50 mg/dl increase in Lp(a) concentration (−125 nmol/l) was associated with a 31% increase in the risk of death from any cause and from ASCVD in the general population and 15% in patients with ASCVD [49].
Lp(a) is recognised as a strong risk factor for the development of AVS, promotes aortic valve calcification (CAVS) and is associated with the progression of CAVS as confirmed in several studies [47, 50–52]. Available research indicates that Lp(a) influences the risk of developing AVS, even in patients with Lp(a) levels between 20 and 64 mg/dl (−50–160 nmol/l) (RR = 1.6, 95% CI: 1.1–2.4), while those with levels > 90 mg/dl (> 225 nmol/l) show an almost threefold increase in risk [37]. A meta-analysis involving 8 studies and 52,931 participants found that plasma Lp(a) levels ≥ 50 mg/dl (≥ 125 nmol/l) were associated with a 1.76-fold increased risk of CAVS (RR = 1.76; 95% CI: 1.47–2.11) [53]. An analysis of two prospective cohorts including a total of 145 patients with CAVS, showed that those with Lp(a) levels > 35 mg/dl (> 87.5 nmol/l) had faster disease progression, as well as a higher risk of the composite endpoint of aortic valve replacement and death [52].
Polish and international data indicate that the ASCVD population, which is least effectively treated and monitored for lipid disorders are patients with peripheral artery disease. Unfortunately, elevated Lp(a) levels also further increase this risk of PAD and its complications. In a study involving 1,472 patients with PAD, participants with Lp(a) levels > 30 mg/dl (> 75 nmol/l) had a higher risk of PAD-related surgery (HR = 1.20, 95% CI: 1.02–1.41) and lower limb peripheral revascularisation (HR = 1.33, 95% CI: 1.06–1.66]) [54]. In a systematic review including 15 studies and 493,650 patients, most studies confirmed a significant association between high Lp(a) levels and the risk of PAD. High Lp(a) levels have also been found to be associated with increased PAD progression, risk of intermittent claudication, recurrence of significant stenosis and risk of hospitalisation and death [55]. Higher levels of Lp(a) have been shown to be independently associated with an increased risk of major adverse limb events (MALE) [56]. A recently published analysis of pooled data from two randomised clinical trials TRA2P-TIMI 50 and FOURIER involving patients with stable atherosclerotic vascular disease confirms a significant association of Lp(a) with progression of lower extremity arteriosclerosis (PAD) [57].
The association between Lp(a) and HF is highly dependent on the effect of Lp(a) on coronary artery disease (CAD) and CAVS. Results from a large case-control study involving 143,087 participants confirmed the association of LPA genetic variants with HF risk; each 50 nmol/l (~20 mg/dl) increase in concentration was associated with a 5% increase in the risk of developing HF [58].
The role of Lp(a) in the development of atrial fibrillation (AF) is also not fully established. An analysis of UK biobank data involving 435,579 participants showed that each increase in Lp(a) of 50 nmol/l (−20 mg/dl) was associated with a 3% increase in the risk of AF [59]. Although coronary artery disease (CAD) itself is a risk factor for the occurrence of AF, according to some authors, Lp(a) may influence the development of AF independently of ASCVD [60, 61]. A meta-analysis including 275,647 AF cases and 2.1 million patients with Lp(a) showed that increased Lp(a) was associated with a very small increased risk of AF in studies based on Mendelian randomisation (OR = 1.024, 95% CI: 1.007–1.042, p < 0.001). The authors found a higher risk of AF in the European cohort (OR = 1.023, 95% CI: 1.007–1.040, p < 0.001) and a low risk (OR = 0.940, 95% CI: 0.893–0.990) in the Chinese population [4]. On the other hand, Garg et al. noted an inverse relationship between Lp(a) concentration and AF incidence after a follow-up period of almost 13 years [62]. A recent systematic review covering 11 observational studies and almost 1.25 million patients did not conclusively confirm the usefulness of Lp(a) assessment as a marker of AF risk in clinical practice and highlighted the need for further studies in this area [63].
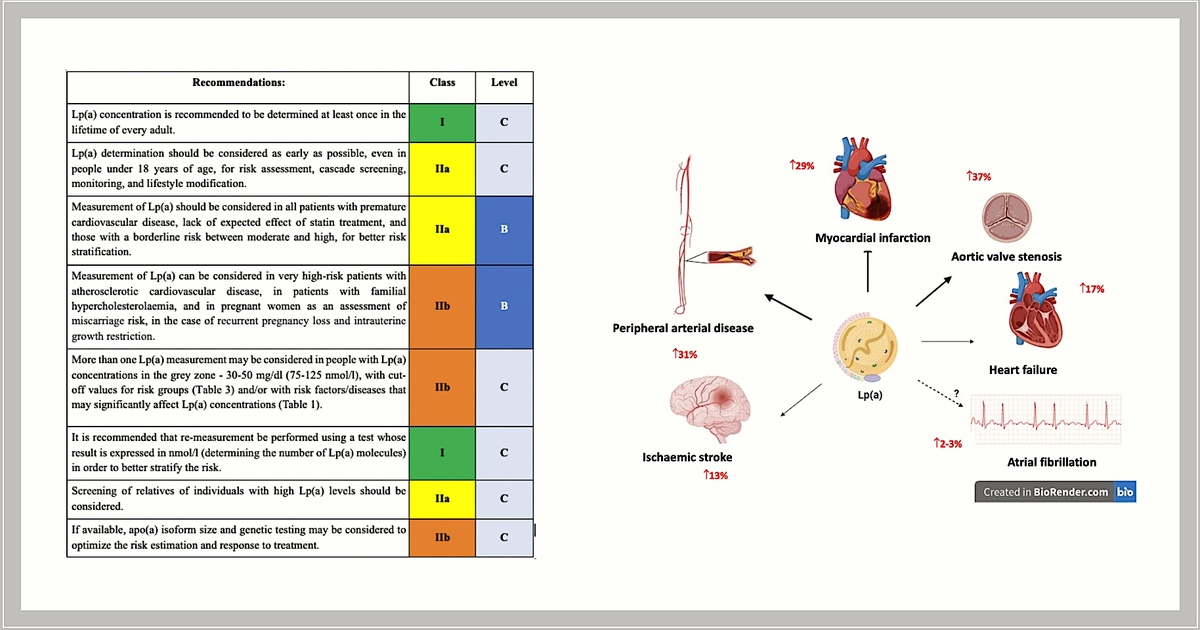
Expert opinion: Elevated lipoprotein(a) concentrations are an independent (residual) risk factor for cardiovascular disease and Lp(a) should be determined at least once in every person’s lifetime (IA). Available data indicate a strong causal relationship between elevated Lp(a) concentrations and an increased risk of cardiovascular disease and its complications, peripheral artery disease, ischaemic stroke (by up to 30% for each increase in concentration of 20–50 mg/dl [50–125 nmol/l]), as well as a very high risk of developing aortic valve stenosis (up to three times higher for high Lp(a) concentrations).
Lp(a) concentration, genetic factors
Blood Lp(a) levels range from < 0.1 mg/dl to > 300 mg/dl (< 0.2 – > 750 nmol/l) and are approximately 90% genetically determined [64]. The main genetic factor determining blood Lp(a) levels is (apo(a) isoform size-dependent) the differential number of copies associated with the number of coding kringle-IV (K-IV) repeats in the LPA gene. The number of K-IV type 2 domain repeats determines the size of the apo(a) isoforms and is inversely proportional to Lp(a) concentration [65]. A low number of K-IV type 2 domain repeats (≤ 22) characterises small apo(a) isoforms and is associated with higher Lp(a) concentrations compared to large apo(a) isoforms [16]. However, the exact relationship between apo(a) isoform and Lp(a) concentrations is complex rather than linear and is also modified by functional single nucleotide polymorphisms (SNPs) [66]. Two well-known SNPs affecting Lp(a) concentrations are rs10455872 and rs3798220 usually observed for small apo(a) isoforms and indicative of high Lp(a) concentrations [39].
At birth, Lp(a) concentration is low and increases with age until about 5 years of age, when it usually reaches the value that is determined in adulthood [67]; however, it has been shown that it can continue to rise until adulthood [68, 69]. Lp(a) concentrations are influenced by race and gender. Lower Lp(a) concentrations are found in Chinese (mean 16 nmol/l [−6.4 mg/dl]), Caucasians (19 nmol/l [−7.6 mg/dl]) and South Asians (31 nmol/l [−12.4 mg/dl]), and highest in Black people (75 nmol/l [−50 mg/dl]) [45]. Women of both Caucasian and Black race are observed to have higher Lp(a) levels by approximately 5–10% compared to men [70–72]. Additionally, in women, Lp(a) levels are more variable and usually increase during menopause [73]. A recent study showed a 27% increase in Lp(a) levels in postmenopausal women [71].
Other factors influencing Lp(a) concentrations
Genetic factors have a strong influence on Lp(a) concentrations; however, some non-genetic factors can also modify them. Lifestyle changes have a minimal effect on Lp(a) levels [74]. Results from a recent dietary study show that a low-carbohydrate diet (LCD) rich in saturated fat reduced Lp(a) concentrations by 15%, and decreased insulin and triglyceride levels (after 20 weeks compared with a high-carbohydrate diet) [75]. In another study, the use of a ketogenic diet resulted in a 26% reduction in Lp(a) (after 3 weeks) [76]. However, on the other hand, it has been shown that a ketogenic diet can lead to increased total cholesterol, LDL-C and apoB [77], higher risk of type 2 diabetes [78] and, consequently, higher cardiovascular and all-cause mortality [79]. Hence, studies to date are not sufficient to consider a low-carbohydrate or ketogenic diet in people with elevated Lp(a) [80].
Reducing saturated fatty acids (SFA) intake, which is associated with lower LDL-C and reduced CVD risk, resulted in an increase in Lp(a) [81]. The inverse effect of dietary SFA reduction on LDL-C and Lp(a) [82], despite the lack of data on the clinical significance of this discrepancy, raises some questions regarding dietary recommendations [83]. It seems likely that the increase in Lp(a) concentrations as a result of SFA reduction may play an underestimated role as a determinant of CVD risk in individuals with very high Lp(a) concentrations and elevated LDL-C concentrations [83]. Therefore, the approach to dietary recommendations should be personalised [84].
Physical activity, increasing physical exertion, such as intensive strength training, or increasing cardiorespiratory fitness, has very little or no effect on Lp(a) concentrations, whereas it significantly affects the concentrations of other lipids and lipoproteins, especially triglycerides and HDL-C, and only has an impact on LDL-C when accompanied with weight loss [85]. A minimal modulating effect of exercise on Lp(a) levels has been reported in children and adults under 35 years of age and in diabetic patients, but this needs to be confirmed in further studies [74].
Endogenous sex hormones have little or no effect on Lp(a) levels [73] although recent data indicate an increase in Lp(a) concentrations in menopausal women. Post-menopausal hormone replacement therapy, on the other hand, reduces Lp(a) concentrations by 20-25% [86]. During pregnancy, Lp(a) values increase ~2-fold and return to baseline levels after delivery [87], hence some available studies have indicated an increased risk of pregnancy complications, including preeclampsia, low birth weight or preterm delivery, and some guidelines recommend monitoring Lp(a) levels during pregnancy [88]. However, the data are inconclusive, and a recent Mendelian Randomisation study does not appear to support a causal relationship between Lp(a) levels and the risk of preeclampsia [89].
Growth hormone treatment in adults with growth hormone deficiency results in a 2-fold increase in Lp(a) concentration [90, 91]. Lp(a) levels are decreased in hyperthyroidism and increased in hypothyroidism [92]. Treatment of overt hyperthyroidism with thyroxine or radioactive iodine therapy increased Lp(a) concentrations by 20–25% [92]. Levothyroxine in overt and subclinical hypothyroidism slightly reduces Lp(a) levels [93].
Kidney diseases increase Lp(a) levels, which start to rise even in the early stages of kidney damage [94]. Nephrotic syndrome causes a 3–5-fold increase [95] (compared to controls), and a 2-fold increase in Lp(a) levels has been observed in peritoneal dialysis patients (compared to controls) [96]. Kidney transplantation normalises Lp(a) levels to baseline values [97].
The concentration of Lp(a) depends on the synthesis of apo(a), which takes place in liver cells, hence Lp(a) concentrations decrease with the progression of liver damage [98, 99] (Table I [100–104]).
Expert opinion: Genetic factors are responsible for approximately 90% of Lp(a) concentrations. The remaining approx. 10 % can be influenced mainly by diet, hormonal disorders or kidney and/or liver disease. There are differences in Lp(a) concentrations between men and women (Lp(a) values are on average up to 10% higher in women, with an additional increase after menopause of up to 30%), depending on race, and to a lesser extent on age. Apart from general prevention principles aimed at changing lifestyle and optimising treatment of comorbidities, there are no additional management recommendations in the context of nongenetic factors for Lp(a).
Table I
Non-genetic factors and their influence on Lp(a) concentrations. Modified based on [100] completed with the information from: 1[101]; 2[102]; 3[103]; 4[104]
Who should be tested for Lp(a) and when
The most recent guidelines from the European Atherosclerosis Society (EAS) and the American Heart Association (AHA) recommend that every adult should have their Lp(a) levels determined at least once in their lifetime to identify individuals with very high congenital Lp(a) levels, which significantly increase cardiovascular risk [100, 105] (Table II). It seems that, similarly to the determination of a complete lipid profile in a 6-year-old’s health check-up, which is likely to become possible in Poland in the near future to allow early diagnosis of familial hypercholesterolaemia (FH), Lp(a) testing in childhood also seems reasonable, even though we still essentially lack effective management (beyond monitoring) to successfully lower Lp(a) as early as possible (and studies showing significant benefits of such an intervention).
Table II
Recommendations for lipoprotein(a) testing
Hence, for the time being, it is particularly important to determine the level of this lipoprotein in patients with premature cardiovascular disease, in the absence of the intended effect of statin treatment (data indicate that elevated Lp(a) levels can be expected in patients with little or no response to statin treatment [106]), and in those with a borderline risk between moderate and high, for better risk stratification. Measurement of Lp(a) should also be considered in very high cardiovascular risk patients with ASCVD, in patients with FH, and in pregnant women with recurrent pregnancy loss and intrauterine growth restriction as prevention of miscarriage [88].
According to available data, a single accurate measurement of molar Lp(a) concentration appears to be sufficient to estimate cardiovascular risk [107]. However, repeated measurements of the Lp(a) level are recommended in patients whose Lp(a) concentration in the first measurement was 30–50 mg/dl (75–125 nmol/l), i.e. a value in the so-called grey zone or close to the cut-off points for the risk groups (Table III; possibly resulting in a change in the cardiovascular risk of the given patient); ideally, the repeated measurement should be performed using a test whose result is expressed in nmol/l (defining the number of Lp(a) molecules) for better risk stratification [108]. Repeat measurements of Lp(a) may also be considered in patients who have developed chronic kidney disease, particularly nephrotic syndrome, as such conditions can cause a significant increase in Lp(a) [95]. In addition, in women, it is recommended that Lp(a) be re-measured after the age of 50 [73] (Tables I and II).
Table III
Intervention strategies as a function of total cardiovascular risk and untreated Lp(a) concentration. This table is for reference purposes only, showing that at high Lp(a) values, the risk of a cardiovascular event is significantly underestimated. It also shows that modification of risk factors, such as elevated LDL-C and/or blood pressure, can reduce a person’s overall risk, even if the risk attributable to Lp(a) remains unchanged. To assess the risk associated with elevated Lp(a) and the effects of interventions that reduce Lp(a) and other risk factors, use the calculator available at: http://www.lpaclinicalguidance.com
| Total CV risk excluding Lp(a) (Pol-SCORE) %. | Lp(a) concentration | ||||||
|---|---|---|---|---|---|---|---|
| < 10 mg/dl (< 25 nmol/l) | 10–30 mg/dl (25–75 nmol/l) | 20–50 mg/dl (75–125 nmol/l) | 50–75 mg/dl (125–188 nmol/l) | 75–100 mg/dl (188-250 nmol/l) | ≥ 100 mg/dl (≥ 250 nmol/l) | ||
| Primary prevention | < 1, low risk | Lifestyle changes | Lifestyle changes | Lifestyle changes | Lifestyle changes | Lifestyle changes, pharmacological treatment should be considered* | Lifestyle changes along with concurrent pharmacotherapy |
| ≥ 1 to < 5 or risk moderate | Lifestyle changes | Lifestyle changes | Lifestyle changes | Lifestyle changes, pharmacological treatment should be considered | Lifestyle changes, pharmacological treatment should be considered | Lifestyle changes along with concurrent pharmacotherapy | |
| ≥ 5 to < 10 or risk large | Lifestyle changes | Lifestyle changes | Lifestyle changes, pharmacological treatment should be considered | Lifestyle changes along with concurrent pharmacotherapy | Lifestyle changes along with concurrent pharmacotherapy | Lifestyle changes along with concurrent pharmacotherapy | |
| ≥ 10 or risk very large | Lifestyle changes | Lifestyle changes, pharmacological treatment should be considered | Lifestyle changes along with concurrent pharmacotherapy | Lifestyle changes along with concurrent pharmacotherapy | Lifestyle changes along with concurrent pharmacotherapy | Lifestyle changes along with concurrent pharmacotherapy | |
| Secondary prevention | Very high risk | Lifestyle changes, pharmacological treatment should be considered | Lifestyle changes along with concurrent pharmacotherapy | Lifestyle changes along with concurrent pharmacotherapy | Lifestyle changes along with concurrent pharmacotherapy | Lifestyle changes along with concurrent pharmacotherapy | Lifestyle changes along with concurrent pharmacotherapy |
* Pharmacotherapy refers to the optimal treatment of risk factors, related to LDL cholesterol levels, blood pressure, glucose levels. Prepared on the basis of [100].
These recommendations are particularly important because recent results from the OCEAN (a)-Dose study indicate significant long-term individual variability in Lp(a) levels in patients with ASCVD and elevated baseline Lp(a) concentrations, suggesting that it may be necessary to measure this parameter more than once in a lifetime [109]. The study authors observed as much as 10% (SD 3.9%) mean variation in Lp(a) concentration. The absolute differences observed over time in the placebo group at all visits averaged 22 ±21 nmol/l (9 ±8.5 mg/dl), with a maximum absolute difference of up to 135 nmol/l (54 mg/dl).
According to the European Atherosclerosis Society (EAS) consensus published in 2022 and the American Heart Association (AHA) guidelines, screening of relatives of people with high Lp(a) levels is recommended [100, 105]. A recently published UK Biobank cross-sectional study confirmed that the effectiveness of cascade screening of first-degree relatives of people with high Lp(a) levels, is over 40% [110]. Screening is also recommended in young people with a history of ischaemic stroke or a family history of early ASCVD or high Lp(a), without other identifiable risk factors. Cascade testing for high Lp(a) is recommended in cases of familial hypercholesterolaemia, a family history of high Lp(a) and personal or family history of ASCVD [100].
How often is the Lp(a) concentration tested?
Elevated Lp(a) levels > 50 mg/dl (> 125 nmol/l) are estimated to occur in more than 1.4 billion people worldwide [9]. In Europe, approximately 20% of women and 20% of men have Lp(a) levels > 50 mg/dl (> 125 nmol/l) [88,100], and up to >30% of patients with ASCVD and/or FH may have elevated Lp(a), however, Lp(a) levels are still unfortunately not commonly determined [111]. The EAS, as part of the Lipid Clinic Network (LCN) project, conducted a survey to find out how widespread routine testing of Lp(a) levels is. The survey was completed by 151 clinicians and 75.5% of the respondents declared that they routinely measure Lp(a) in clinical practice. However, across all the European centres surveyed, the results varied widely; in Western European countries, ~90% of clinicians reported that they routinely order Lp(a) measurements, while in Central and Eastern European countries, the figure was ~49% [10]. It is also worth noting that these data are definitely overestimated, as they were conducted among physicians working in specialised lipidology centres.
A study of more than 5 million adults at six academic medical centres affiliated with the University of California in the United States found that the frequency of Lp(a) determination remains very low at 0.3%, with a 4% prevalence among patients with cardiovascular diseases [11]. A study including all adults (4.6 million) registered with the largest health organisation in Israel showed that only 0.1% had their Lp(a) levels tested between 2015 and 2021 [12].
Difficulties in determining Lp(a) concentration
Determination of Lp(a) concentration is not easy, as different results can be obtained depending on the assay chosen, which is partly dependent on the size of the apo(a) isoform [100, 112, 113]. Commercially available tests for the determination of Lp(a) concentrations are immunoenzymatic assays using polyclonal antibodies against apo(a). These assays are more or less sensitive to apo(a) isoforms and therefore the determined Lp(a) concentration can potentially be underestimated for small isoforms or overestimated when large isoforms are present. For accurate determination of Lp(a) concentration, assays based on antibodies directed against the unique apo(a) kringle V domain should be used, which would allow each Lp(a) molecule to be recognised only once [100, 114].
Isoform-sensitive tests report the mass of Lp(a) particles (mg/dl), while isoform-insensitive tests report the molar concentration (nmol/l) of the number of apo(a) particles. The use of a standard factor to convert units from mg/dl to nmol/l is inaccurate and therefore not recommended for use by clinicians [115], although, due to existing differences in the tests used, it is still widely practised (the most common conversion factor is: nmol/l = 2.5 × mg/dl and mg/dl = 0.4 × nmol/l). Recent results from a sub-analysis of the ODYSSEY Outcomes study with alirocumab, in which Lp(a) was measured with three different assays (using immunoenzymatic mass assays, molar assays and molar mass spectrometry-based assays), showed a similar risk reduction regardless of the Lp(a) concentration measurement used [116].
Recently, a mass spectrometric method has been developed and validated for the standardisation of Lp(a) measurements, which addresses the problem of size polymorphisms by selecting specific peptides for quantification that are not present in the KIV2 region [117]. Research is also currently underway on the global standardisation of Lp concentration measurements, which will improve diagnostic interpretation and clinical decision-making regarding Lp(a)-dependent CVD risk [117, 118].
Which concentrations indicate increased cardiovascular risk?
The relationship between Lp(a) concentration and cardiovascular risk is linear, meaning that the risk is higher with increasing Lp(a) concentration [45]. There is a need on the part of clinicians to define a threshold Lp(a) concentration at which cardiovascular risk increases. The Lp(a) level that requires appropriate clinical intervention is ≥ 30 mg/dl (≥ 75 nmol/l) (Table IV) [100]. Individuals whose Lp(a) level is between 30 and 50 mg/dl (75–125 nmol/l) belong to the so-called grey zone, which depends both on the accuracy of the test used to assess Lp(a) levels, but also on all risk factors of the individual patient [100]. However, these individuals already have an increased risk of adverse effects and should be classified as being at moderate risk. High risk is predicted for all patients with Lp(a) concentrations > 50 mg/dl (> 125 nmol/l) and actually for the concentration range 50–180 mg/dl (125–450 nmol/l), while very high risk for values > 180 mg/dl (450 nmol/l).
Table IV
Classification of Lp(a) concentrations according to cardiovascular risk category. Modified from [88]
How to stratify risk
Widely used cardiovascular risk assessment tools for primary prevention do not take Lp(a) levels into account. In 2021, the Polish guidelines of six scientific societies for the management of lipid disorders for the first time used an elevated Lp(a) concentration > 50 mg/dl (125 nmol/l) as an additional parameter defining extremely high cardiovascular risk in a patient after an acute coronary syndrome (ACS) with diabetes [88]. If the Lp(a) concentration is high but not included in the risk calculation, then the calculated risk of CVD events is significantly underestimated [43]. Based on an analysis of UK biobank data, the EAS consensus showed that people with Lp(a) levels of 30 (75 nmol/l), 50 (125 nmol/l), 75 (187.5 nmol/l), 100 (250 nmol/l) and 150 mg/dl (375 nmol/l) compared to those with an average Lp(a) concentration of 7 mg/dl (17.5 nmol/l) have an increased cardiovascular risk of 1.22; 1.40; 1.65, 1.95 and 2.72, respectively. Thus, if the baseline risk is 25% and in addition there is a high Lp(a) level of e.g. 150 mg/dl (375 nmol/l) then the risk increases from 25% to 68% [43, 100].
The EAS, in its 2022 recommendations, switched to a new and extremely useful risk assessment calculator that takes into account Lp(a) levels in combination with traditional cardiovascular risk factors (Table III) (http://www.lpaclinicalguidance.com/) [43, 100]. This calculator estimates the risk of myocardial infarction or stroke up to the age of 80 years, with or without consideration of Lp(a) levels.
Currently available therapies affecting Lp(a) levels
Currently available lipid-lowering medicines have an effect on Lp(a) concentrations, but their efficacy in this respect varies and is mostly insufficient (Table V).
Table V
Effect of currently available hypolipemic therapies on Lp(a) levels
| Intervention | Effect on Lp(a) levels |
|---|---|
| Statins | Possible ↑ by 6–10% |
| Ezetimibe | ↓ Up to 7% |
| Bempedoic acid | No effect |
| Alirocumab | ↓ 25–35% |
| Evolocumab | ↓ 25–35% |
| Inclisiran | ↓ 20–25% |
| Niacin | ↓ 30–40% |
| Lp(a) apheresis | ↓ 25–40% |
In the case of statins, there are conflicting findings regarding their effect on Lp(a) levels. Some studies have shown that statins are effective in lowering Lp(a) levels [119, 120], while others have indicated an increase in Lp(a) of 10–20% [121, 122]. There are also studies showing no effect of statins on Lp(a) levels [123]. Besides, the effect on Lp(a) may vary depending on the type of statins [124]. The discrepancy in study results may be explained by the fact that statins affect Lp(a) levels differently in dyslipidaemic patients, depending on the apo(a) phenotype. Statins only increase Lp(a) levels in patients with a low molecular weight (LMW) apo(a) phenotype (≤ 22 KIV), and in these individuals a mean absolute increase of up to > 30 mg/dl (> 40%) can be observed [125].
Statin-induced increases in Lp(a) levels are not clinically relevant and there are no recommendations to discontinue statin treatment in patients with high Lp(a) levels [43, 88, 100, 125]. A meta-analysis of 20 randomised clinical trials involving 23,605 participants showed that atorvastatin showed a slightly worse effect on Lp(a) levels compared to other statins, whereas pitavastatin could potentially reduce Lp(a) levels [126]. This was also confirmed by the results of the VISION study, indicating no effect of pitavastatin on Lp(a) and even a numerical reduction in Lp(a) levels [127]. A possible change from statin to pitavastatin in patients with elevated Lp(a) needs to be confirmed in further studies. In the just-published recommendations of Polish experts, they indicate the possibility of considering pitavastatin in patients with elevated Lp(a) (IIb) concentrations, while emphasising the need for further studies confirming this relationship, as well as assessing the effect of pitavastatin on the size of apolipoprotein(a) isoforms [128].
Ezetimibe has a neutral effect or may cause a slight reduction in Lp(a) although probably not clinically relevant [129]. Similarly, bempedoic acid has been shown to have no clinically significant effect on Lp(a) concentrations [130]. This does, however, imply, in the case of statin-induced increases in Lp(a) concentrations, that combination therapy with a lower dose of statins may be optimal in this group of individuals, although further studies are needed to corroborate the effect of such treatment on Lp(a) concentrations and risk reduction.
The FOURIER and ODYSSEY OUTCOMES studies showed that PCSK9 inhibitors reduced Lp(a) levels: evolocumab by a mean of 29.5% [131, 132] and alirocumab by a mean of 23.5% [133]. Based on sub-analyses of the FOURIER and ODYSSEY OUTCOMES clinical trials, absolute CVD risk reductions from 2.4% to 3.7% (ARR 1.3%, number needed to treat [NNT] = 77) were demonstrated (as an effect of the reduction of Lp(a) concentration) [134]. Another analysis of the same study suggests that the reduction in Lp(a) resulting from the use of alirocumab may also significantly reduce the risk of PAD [135]. A study that assessed the effect of early initiation of evolocumab therapy on Lp(a) levels after primary percutaneous coronary intervention (PCI) in patients with myocardial infarction showed that early initiation of evolocumab in combination with pitavastatin rapidly inhibited the increase in Lp(a) levels compared with pitavastatin alone. Evolocumab (PCSK9 inhibitors) may partially attenuate statin-induced increases in Lp(a) levels, suggesting its benefit in managing residual risk in patients with elevated Lp(a) levels after myocardial infarction [136]. The reduction in Lp(a) as a result of PCSK9i administration is achieved through a dual mechanism involving both increased catabolism and reduced Lp(a) production [137]. In comparison, available data on therapy based on an RNA interference mechanism, based on the results of the ORION 10 and ORION 11 studies, showed a reduction in Lp(a) by ~20% following the administration of inclisiran [138, 139].
It is noteworthy that apo(a) size is an independent determinant of the response to PCSK9i in terms of Lp(a) reduction. The presence of each additional kringle domain is associated with an increased Lp(a) reduction of 3%; in other words, the larger the size of the dominant apo(a) isoform, the greater the relative Lp(a) reduction. This partly explains the variable efficacy of PCSK9i and allows individualisation of therapy by identifying those patients who will benefit most from Lp(a) reduction [140].
It is worth noting that in the B101 drug programme in Poland there is no indication for the use of PCSK9 protein modulators to reduce Lp(a) levels, just like in most European countries [88, 100]. It is possible to use PCSK9 protein modulators to reduce elevated Lp(a) concentrations if the patient simultaneously meets other applicable programme criteria, or as part of the rescue access to drug technology (RDTL) procedure, which is certainly used definitely too rarely in Poland.
Current data suggest that niacin, which is a medicine unfortunately not available in Poland or Europe, may have a beneficial effect on Lp(a) levels [108]. Niacin can reduce Lp(a) in a dose-dependent manner by an average of 30–40% and by about 20% in those with the highest Lp(a) levels [88, 141]. Niacin can be used in combination with statin therapy. It was shown that in individuals with Lp(a) levels above 300 nmol/l (120 mg/dl), the reduction in Lp(a) during niacin treatment was 63% compared to a 3.9% reduction with statin therapy. Genetic studies revealed a specific variant of the LPA promoter gene that appeared to increase the efficacy of niacin treatment [142]. The effect of niacin on Lp(a), similarly to other lipid-lowering drugs, was also shown to be dependent on the size of the apo(a) isoform [143]. The use of niacin reduced Lp(a) levels by 28% and lipoprotein-associated phospholipase A2 (Lp-PLA2) levels by 22% in the low molecular weight (LMW) apo(a) group, while no significant changes in Lp(a) levels were observed in the high molecular weight (HMW) apo(a) isoform group [144]. Another study that analysed the effect of niacin on Lp(a) found that the largest apo(a) isoform sizes (corresponding to lower Lp(a) levels) resulted in a 50% and 4 nmol/l reduction in Lp(a) in absolute terms, while the smallest apo(a) isoform sizes (corresponding to higher Lp(a) levels) showed a reduction in Lp(a) of 16% and 30 nmol/l in absolute terms [141] – a similar effect is observed for PCSK9 inhibitors. Due to the emergence of studies demonstrating a lack of clinical benefit from the use of niacin and the risk of adverse effects [145–147], current EAS and Polish guidelines do not recommend its use [88, 100].
The results of the above studies suggest that genetic testing and phenotyping should be ordered in patients with elevated Lp(a) levels, which could serve as an additional tool to assess CVD risk, to personalise and select the best available therapy (statins, niacin, PCSK9 inhibitors) [142, 143]. However, currently available techniques for the determination of apo(a) isoforms are complex, expensive and their availability is still very limited [148].
Lipoprotein apheresis is an efficient and effective therapeutic option for lowering Lp(a) levels [149, 150]. A single apheresis treatment reduces Lp(a) by approximately 60–75%, and regular treatments every 1–2 weeks reduce Lp(a) by approximately 25–40% compared to baseline values [151–153]. Studies have shown that the use of lipoprotein apheresis in patients with ASCVD and elevated Lp(a) levels results in a significant reduction in CVD events. A prospective multicentre study of 170 patients with ASCVD and Lp(a) > 60 mg/dl (> 150 nmol/l) showed a significant reduction in the annual rate of MACE after implementation of apheresis therapy [154]. Data from the German Lipoprotein Apheresis Registry (GLAR) in ASCVD patients with elevated Lp(a) levels showed a 72.4% reduction in Lp(a) and a significant reduction in the incidence of ASCVD events (> 80%) over 7 years, regardless of baseline LDL-C levels [155]. A US study observed a 63% reduction in Lp(a) and a 94% reduction in major adverse cardiovascular events (MACE) over a mean lipoprotein apheresis treatment period of 48 months [156]. A study in Polish patients showed a 73% reduction in Lp(a) levels and a significant reduction in CV incidents [157]. In addition, an additional effect of changes in the fatty acid profile was observed [158]. In a randomised trial in patients with refractory angina and Lp(a) > 50 mg/dl (> 125 nmol/l), lipoprotein apheresis improved myocardial perfusion on MRI, physical performance in the 6-minute walk test and quality of life [159]. However, there has not yet been a randomised trial evaluating the use of lipoprotein apheresis in patients with high Lp(a) levels, targeting cardiovascular incident analysis [160].
Lipoprotein apheresis is the first treatment for elevated Lp(a) to receive initial Food and Drug Administration (FDA) approval for use in patients with Lp(a) > 60 mg/dl (> 150 nmol/l) regardless of baseline LDL-C levels [160]. Lipoprotein apheresis may be considered in patients with very high Lp(a) levels and progressive ASCVD despite optimal treatment of other risk factors [100]. Apheresis is a safe, effective method; however, it is invasive, costly and of limited availability. The most common (relatively rare – 3–6% and mostly mild to moderate) side effects include puncture site lesions (mainly peripheral veins), transient hypotonia and hypocalcaemia [161].
Mipomersen (antisense oligonucleotide [ASO] – targeting apo(B) mRNA) is able to lower Lp(a) in the range of 21–27% [162–164], but its safety profile limits its use to people with homozygous FH and it is not suitable for routine administration to lower Lp(a) levels [165]. This medicine is not available in Poland.
Current therapies used to lower LDL-C have a moderate effect on Lp(a). Reducing the risk of major adverse cardiovascular events may require greater reductions in Lp(a) with more potent and targeted therapies which are still undergoing clinical trials (Tables VI–VIII).
Table VI
Recommendations for the management of elevated Lp(a) concentrations
Table VII
Practical recommendations for the management of patients from different risk groups with elevated Lp(a) levels > 50 mg/dl (> 125 nmol/l).1 Treatment based on a fixed-dose combination tablet (FDC) is preferred2 for both primary and secondary prevention3 under the B101 drug programme or the rescue access to drug technology (RDTL) procedure
Table VIII
New Lp(a)-lowering medicines under clinical development
Management of elevated Lp(a) concentrations
Currently, there are no approved medicines targeting the lowering of Lp(a) concentrations, and therefore, with elevated Lp(a) values, the recommended management is aimed at lowering overall risk through lifestyle changes, and intensive treatment or optimisation of treatment of other risk factors that can be effectively treated according to current recommendations (Figure 3 and Table V) [88, 100]).
Figure 3
Recommended management pathway for patients with elevated Lp(a) levels. Modified from [108]
CVD – cardiovascular, Lp(a) – lipoprotein(a), RDTL – rescue access to drug technology.
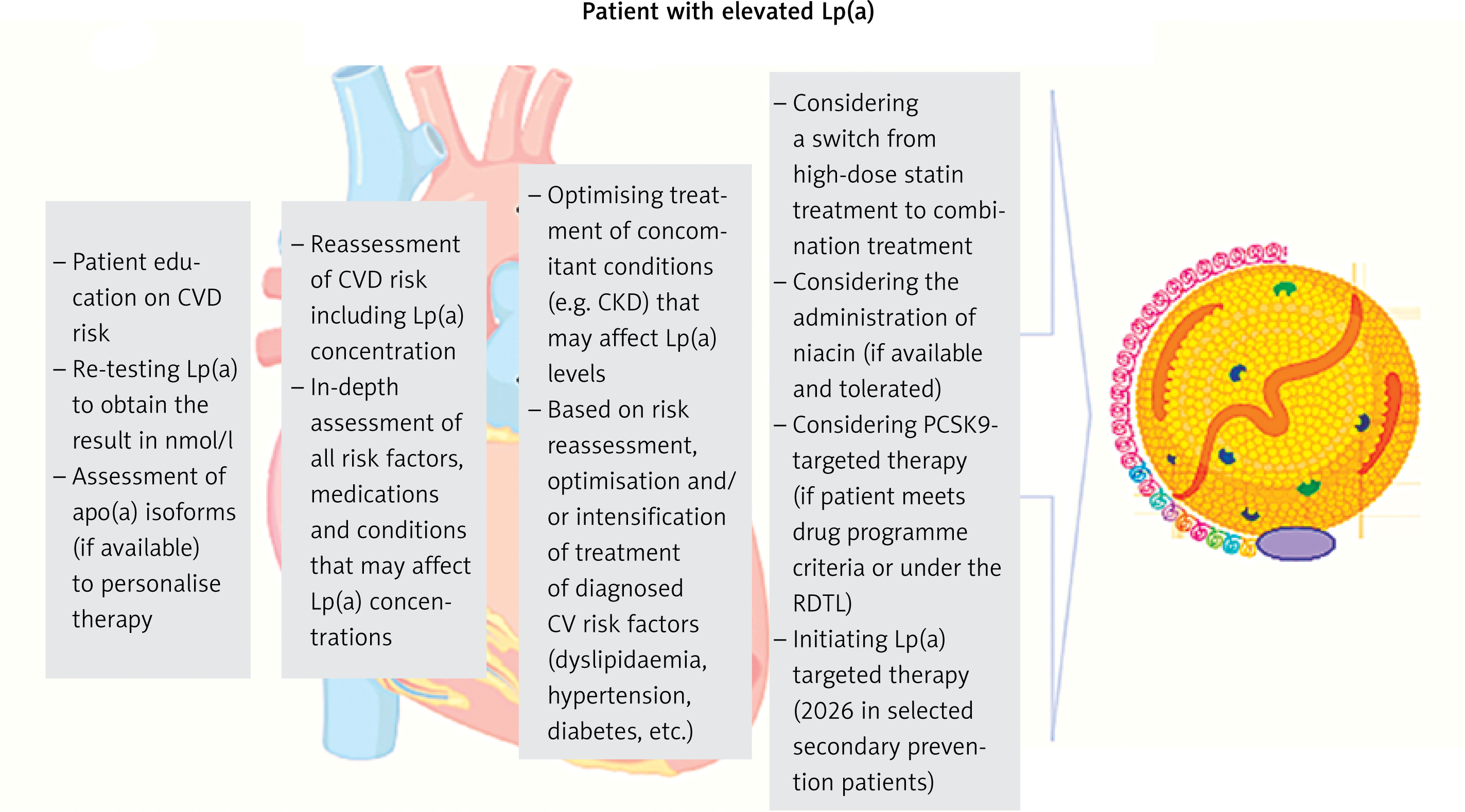
Lifestyle changes have a minimal effect on Lp(a), but significantly affect overall cardiovascular risk, optimising blood pressure and concentrations of lipids and glucose, significantly reducing long-term ASCVD risk [88, 166].
A small increase in Lp(a) after treatment with statins does not seem to be clinically relevant. According to recent recommendations by Polish experts, a change from statin to pitavastatin can be considered in patients with elevated Lp(a), but further studies are still needed to confirm this effect, especially in terms of the impact on cardiovascular risk reduction. In the case of elevated Lp(a) values, a change from statin treatment to a combination treatment of statins with available medicines – ezetimibe or a combination of ezetimibe and bempedoic acid (to be available from 2024 in Poland) and a combination treatment based on inhibition of the PCSK9 protein should be considered (Tables VI and VII) [167]. Lipoprotein apheresis may be considered in patients with very high Lp(a) levels (≥ 60 mg/dl [≥ 150 nmol/l]) and progressive ASCVD, despite optimal treatment of other cardiovascular risk factors [100] (Tables VI, VII).
Management of asymptomatic patients with high Lp(a) levels involves a recommendation of changes in modifiable risk factors through lifestyle changes and treatment according to guidelines. Assessment of the coronary artery calcification (CAC) score can be considered as early as in patients at 45–50 years of age, a low score, or zero, helps to reduce patient concerns about elevated Lp(a) levels. A family history of (premature) ASCVD should be checked, and family screening for high Lp(a) should be suggested. For some patients, referral to a lipid clinic or outpatient specialist care (AOS) should be considered [88, 100].
Future targeted Lp(a) lowering therapies
The strong association between Lp(a) and cardiovascular risk has contributed to the development of new medicines aimed at reducing Lp(a) levels (Table VIII) [168]. Medicines targeting Lp(a) reduction are mainly RNA-based therapies, including antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs), which allow selective silencing of the LPA gene. The mechanism of action of ASOs and siRNAs is to reduce Lp(a) levels by inhibiting apo(a) protein synthesis.
Pelacarsen, a second-generation antisense oligonucleotide, is covalently bound to N-acetylgalactosamine (GalNAc), which provides uptake via asialoglycoprotein receptors located on hepatocytes, thus improving the potency and safety of the medicine. Previous studies have shown that the medicine was well tolerated and resulted in a 72% and 80% reduction in Lp(a) levels, depending on the dose of 60 mg every 4 weeks and 20 mg every week, respectively [169]. The reduction in Lp(a) was independent of LPA genetic variants and isoform size [170]. Currently, pelacarsen, administered at a dose of 80 mg administered subcutaneously once a month, is in a phase 3 clinical trial determining the effect of the medicine on reducing the risk of cardiovascular events (Lp(a)HORIZON trial, NCT04023552). Anticipated study completion date is May 2025 [168].
Olpasiran (AMG-890), Zerlasiran (SLN360) and Lepodisiran (LY3819469) are GalNAc-conjugated siRNAs administered subcutaneously. Olpasiran resulted in a dose-dependent reduction in Lp(a) ranging from 71% to 97% [171]. It is currently in a phase 3 clinical trial (Ocean(a) study, NCT05581303) with an expected study completion date of December 2026. The results, which can be expected in 2027, will provide new information on the pharmacokinetic and pharmacodynamic properties of olpasiran, the best dosing and its effect on cardiovascular events.
Zerlasiran at the two highest single doses of 300 mg and 600 mg reduced Lp(a) levels by 96% and 98%, respectively, and plasma Lp(a) levels were still significantly reduced (by 70% and 81%, respectively) 150 days after administration [172]. A randomised, placebo-controlled phase II study (NCT05537571) has recently commenced, enrolling 160 high-risk ASCVD participants with Lp(a) levels above 125 nmol/l (50 mg/dl). The aim of this study is to investigate the safety, efficacy, and tolerability profile of zerlasiran, with an estimated completion date of June 2024 [173].
Lepodisiran is in early-stage trials. Phase I clinical trial (NCT04914546) is now completed, which involved 48 adults without CVD and with Lp(a) levels ≥ 75 nmol/l (or ≥ 30 mg/dl) [174]. After 168 days, the greatest reductions in Lp(a) concentrations were achieved for the subcutaneously administered single dose of 304 mg –96% and 608 mg – 97%. At day 337, the median change in Lp(a) concentration was –94% in the group receiving 608 mg lepodisiran. A phase 2, placebo-controlled study (NCT05565742) is currently underway that will evaluate the efficacy and safety of lepodisiran in 254 participants with Lp(a) levels above 175 nmol/l (70 mg/dl) over 20 months. The study is expected to end in October 2024 [175].
Muvalaplin (LY3473329) is a selective small-molecule inhibitor of Lp(a) synthesis taken orally that inhibits Lp(a) formation by blocking the apo(a)-apo B100 interaction, while avoiding interaction with the homologous protein plasminogen. Results from a phase I study (NCT04472676) showed that it was well tolerated and produced a maximum reduction in Lp(a) of 63% to 65% [176]. A phase II study (KRAKEN trial, NCT05563246) is currently underway to evaluate the efficacy and safety of muvalaplin in adult participants with elevated Lp(a) levels (≥ 175 nmol/l; 70 mg/dl) at high risk of cardiovascular events. The study is scheduled for completion in January 2024 [177].
Obicetrapib is a next-generation selective cholesteryl ester transporter protein (CETP) inhibitor, currently in clinical trials for reducing both LDL-C levels and the incidence of major adverse cardiovascular events. A randomised, double-blind, placebo-controlled trial in dyslipidaemic patients (the ROSE trial, NCT04753606) showed that obicetrapib at oral doses of 5 mg and 10 mg, compared with placebo, as an adjunct to intensive statin therapy (40 mg or 80 mg of atorvastatin or 20 mg or 40 mg of rosuvastatin) resulted in reductions in Lp(a) of 33.8% and 56.5%, respectively [178] (Table VIII).