Introduction
Lung cancer is a malignant tumor causing high human mortality worldwide. About 80% of lung cancers are non-small cell lung cancer (NSCLC). The therapeutic efficacy of lung cancer treatment is poor currently [1]. About 40% of patients with NSCLC present with advanced-stage disease at diagnosis. The 5-year survival rate of NSCLC patients is only about 2% [2]. Thus, it is imperative to find novel therapies to improve the therapeutic efficacy for lung cancer. On the other hand, to elucidate the pathogenesis of lung cancer also helps to develop more effective therapeutic approaches.
The p53 protein is also called a tumor suppressor and is encoded by the TP53 gene in humans; it induces tumor cell apoptosis. Published data indicate that the TP53 gene mutation is found in about half of cancer cells [3]. The suppression of p53 expression was also found in about half of cancer cases [4]. Yet, factors in the suppression of p53 remain to be further understood.
It is reported that Bcl2-like protein 12 (Bcl2L12) can suppress p53 expression in gliomas [5]. Bcl2L12 belongs to the Bcl2 family and has multiple functions in the regulation of cell bioactivities [6]. Bcl2L12 expression is highly expressed in most human gliomas [5]. Expression of Bcl2L12 induces resistance to apoptosis. Bcl2L12 can neutralize caspase-7 and caspase-3 [5], which play important roles in the execution phase of apoptosis [7]. Factors in the regulation of Bcl2L12 remain to be further elucidated.
Previous studies indicate that protease-activated receptor-2 (PAR2) can attenuate cancer cell apoptosis [8]. PAR2 is encoded by the F2RL1 gene in humans. PAR2 acts as a sensor for proteolytic enzymes generated during infection and modulates inflammatory responses [9]. Mast cell-released mediators, such as tryptase, can activate PAR2 to contribute to the inflammatory response [10]. Other factors, including Der P1–3, Pen C 13, factor Xa/VIIa, matriptase, cathepsin S, granzyme A, kallikreins (KLK 2/4/5/6/14), MMP-1, elastase, acrosin, chitinase, bacterial gingipains, and testisin to initiate PAR2 activation, can also activate PAR2 [11, 12]. Based on the information above, we hypothesize that activation of PAR2 may increase Bcl2L12 in lung cancer cells, which may suppress the expression of p53 and attenuate cancer cell apoptosis. To test this, we carried out a cell culture study. The results showed that activation of PAR2 increased the expression of Bcl2L12 in lung cancer cells, which markedly attenuated lung cancer cell apoptosis.
Material and methods
Reagents
The antibodies of Bcl2L12, p53 and Mdm2 were purchased from Santa Cruz Biotech (Santa Cruz, CA). The Annexin V kit, protein G agarose and ChIP kit were purchased from Sigma Aldrich (St. Louis., MO). The reagents for RT-qPCR and Western blotting were purchased from Invitrogen (Carlsbad, CA). The PAR2 active peptide (AP) and control peptide (CP) were provided by Enke Biotech (Shenzhen, China).
Cell culture
Human lung cancer cell lines, p53-carrying A549 cells and LU99 cells (purchased from ATCC), were cultured in DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 0.1 mg/ml streptomycin and 2 mM L-glutamine. The medium was changed in 1 or 2 days. The cell viability was greater than 99% as assessed by trypan blue exclusion assay.
Real time quantitative RT-PCR (RT-qPCR)
The lung cancer cells were collected from the culture after experiments. Total RNA was extracted from the cancer cells with the TRIzol reagents. The cDNA was synthesized with the RNA with a reverse transcription kit following the manufacturer’s instructions. The samples were amplified in a qPCR device (CFX96 Touch Real-Time PCR; Bio-Rad) with the SYBR Green Master Mix. The primers used in the study included Bcl2L12 (AAGATGTCTTCCCTGCTCCC and CAAGTCCACGGGTGAAACAG) and p53 (TGGCCATCTACAAGCAGTCA and GGTACAGTCAGAGCCAACCT). The results were calculated by the method of 2–ΔΔct and presented as fold change compared to controls.
Preparation of cytosolic and nuclear extracts
The lung cancer cells were lysed with a lysis buffer at 4°C for 15 min, and centrifuged at 500 × g for 10 min at 4°C. The supernatant was collected and used as the cytosolic extract. The pellet was added to a nuclear extract buffer and incubated for 15 min at 4°C, followed by centrifugation at 13,000 × g for 10 min at 4°C. The supernatant was collected as the nuclear extract. The protein concentrations were determined by the Bradford method.
Western blotting
Total proteins were extracted from the lung cancer cells collected from related experiments. The protein was quantified with the BCA method, fractioned with sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane. After blocking with skim milk (5%) for 30 min at room temperature, the membrane was incubated with the primary antibodies of interest overnight at 4°C, washed 3 times with Tris-buffered saline Tween 20 (TBST), incubated with the second antibodies of interest (conjugated with peroxidase) for 2 h at room temperature and washed with TBST 3 times. The immune blots on the membrane were developed with enhanced chemiluminescence (ECL). The results were photographed with an imaging device (UVI image station, Cambridge, UK).
RNA interference
The Bcl2L12 gene in lung cancer cells was knocked down by transduction with Bcl2L12 shRNA (or non-specific shRNA, used as a control) carrying lentivirus with a purchased reagent kit following the manufacturer’s instructions. The effects of RNA interference (RNAi) were assessed by Western blotting.
Immunoprecipitation (IP)
The nuclear extracts were precleared by incubating the samples with protein G agarose for 2 h at 4°C. After centrifugation at 13,000 rpm for 5 min, the supernatant was collected and incubated with antibodies of interest overnight at 4°C. The immune complexes were precipitated by incubating with protein G agarose for 2 h at 4°C. The samples were centrifuged at 13,000 rpm for 5 min. The pellet was collected. The immune complexes on the agarose beads were eluted with an eluting buffer and analyzed by Western blotting.
Chromatin immunoprecipitation (ChIP)
The lung cancer cells were collected after related experiments and fixed with 1% formalin for 15 min. The cells were then lysed with a lysing buffer and sonicated to shear the DNA into small pieces (200–500 bp). The samples were precleared and immune precipitated as described in the IP procedures. The DNA was recovered from the samples by reverse crosslinking at 65°C for 4 h and digested with proteinase K for 1 h at 45 °C to remove proteins; the DNA was recovered by phenol/chloroform extraction and ethanol precipitation. The DNA or input was analyzed by qPCR with the following p53 promoter primers: GCCTGGAACACTCTTCTCCT and GGTGGTAGGATTGGGAGGAC. The results are presented as fold change against the input.
Assessment of apoptosis of lung cancer cells
To characterize the effect of Bcl2L12 on cisplatin-mediated apoptosis, an Annexin V kit and propidium iodide were used in A549 and LU99 cells after cisplatin treatment (10 μg/ml, 24 h). The cells were analyzed with a flow cytometer (FACSCanto II, BD Bioscience). The data were analyzed by flowjo.
Results
Activation of proteinase-activated receptor-2 increases p53 expression in lung cancer cells
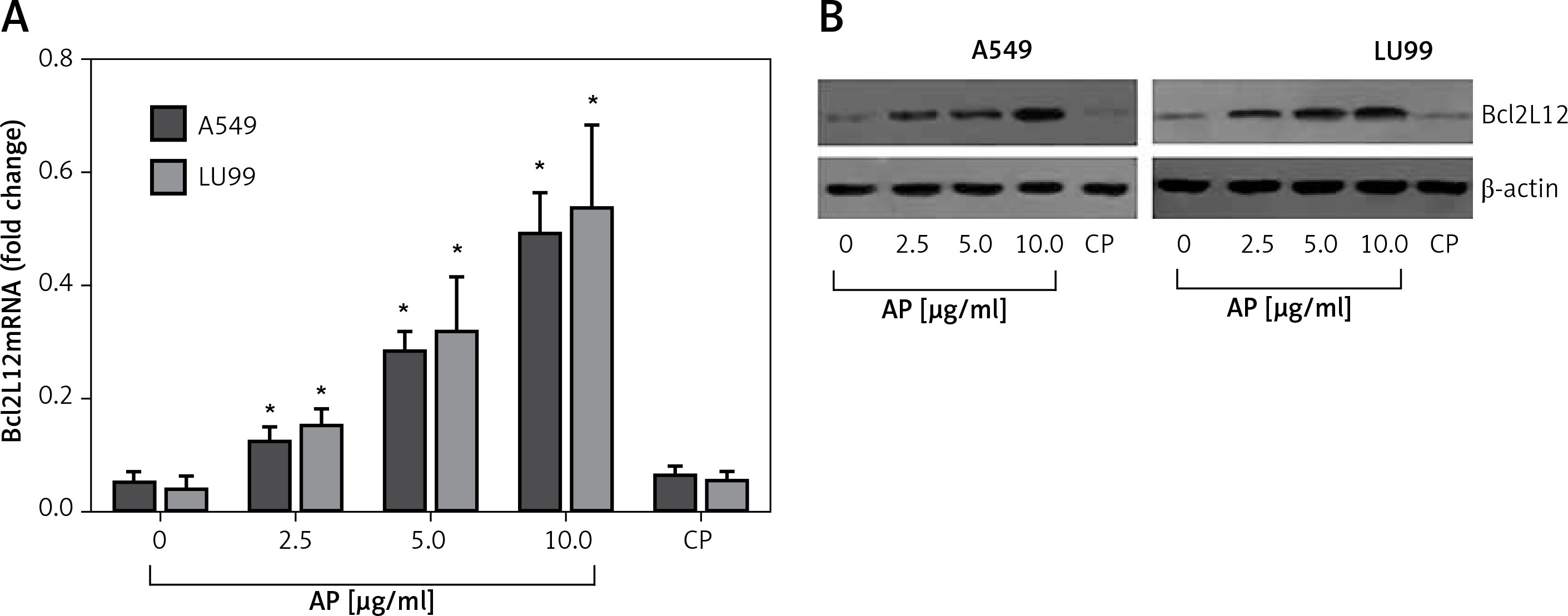
Published data indicate that lung cancer cells express PAR2 [13]. We wondered if activation of PAR2 could regulate the expression of Bcl2L12 in lung cancer cells. To test this, we stimulated the wild type p53 carrying lung cancer cell lines, A549 cells and LU99 cells [14], with active PAR2 peptide (AP) or the control peptide (CP) in the culture for 48 h. The results showed that activation of PAR2 significantly increased the expression of Bcl2L12 in the lung cancer cells in an AP dose-dependent manner. Exposure to CP did not induce any appreciable expression of Bcl2L12 in lung cancer cells (Figure 1). The results demonstrated that activation of PAR2 can enhance the expression of Bcl2L12 in lung cancer cells.
Figure 1
Activation of proteinase-activated receptor-2 (PAR2) increases expression of Bcl2-like protein-12 (Bcl2L12) in lung cancer cells. Levels of mRNA (A) and protein (B) of Bcl2L12 in lung cancer cells after exposure to active peptide of PAR2 (AP) or control peptide (CP) in the culture for 48 h. The data represent 3 independent experiments
*p < 0.01, compared to the dose 0 group.
Bcl2-like protein-12 mediates the effects of proteinase-activated receptor-2 on suppression of p53 expression in lung cancer cells
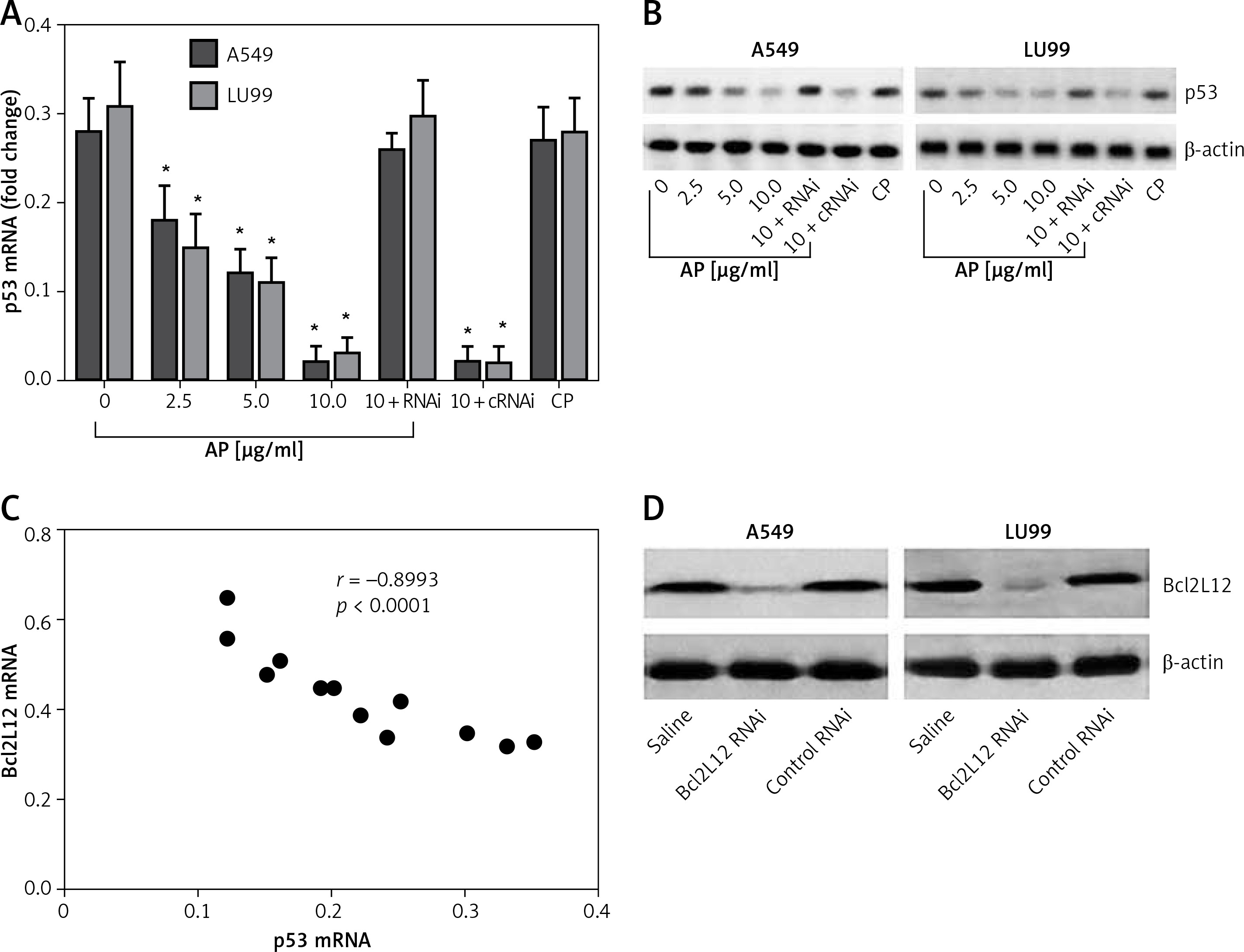
Previous reports indicated that activation of PAR2 can promote cancer cell survival via suppressing p53 expression [15]. In this study, we further tested whether Bcl2L12 was involved in the PAR2-suppressed p53 expression. With the samples collected from the experiments of Figure 1, we analyzed the expression of p53 in the lung cancer cells. The results showed that exposure to PAR2 AP also suppressed the expression of p53 in lung cancer cells (Figures 2 A, B). A correlation assay was performed with the data of Bcl2L12 and p53. A negative correlation was identified between the expression of Bcl2L12 and p53 (Figure 2 C). To corroborate the results, we knocked down the Bcl2L12 gene from the lung cancer cells by RNAi (Figure 2 D). The Bcl2L12-deficient lung cancer cells were exposed to PAR2 AP in the culture for 48 h. Indeed, the inhibitory effects on the p53 expression were abolished (Figures 2 A, B).
Figure 2
Bcl2-like protein-12 (Bcl2L12) mediates the effects of proteinase-activated receptor-2 (PAR2) on suppression of p53 in lung cancer cells. Levels of mRNA (A) and protein (B) of p53 in lung cancer cells (A549 and LU99) after exposure to PAR2 AP (denoted on the X axis of B) for 48 h. C – Correlation between the mRNA of p53 and Bcl2L12. D – The results of Bcl2L12 RNAi
RNAi – lung cancer cells were treated with shRNA of Bcl2L12, cRNAi – lung cancer cells were treated with control shRNA.
Bcl2-like protein-12 forms a complex with Mdm2 to inhibit p53 expression in lung cancer cells
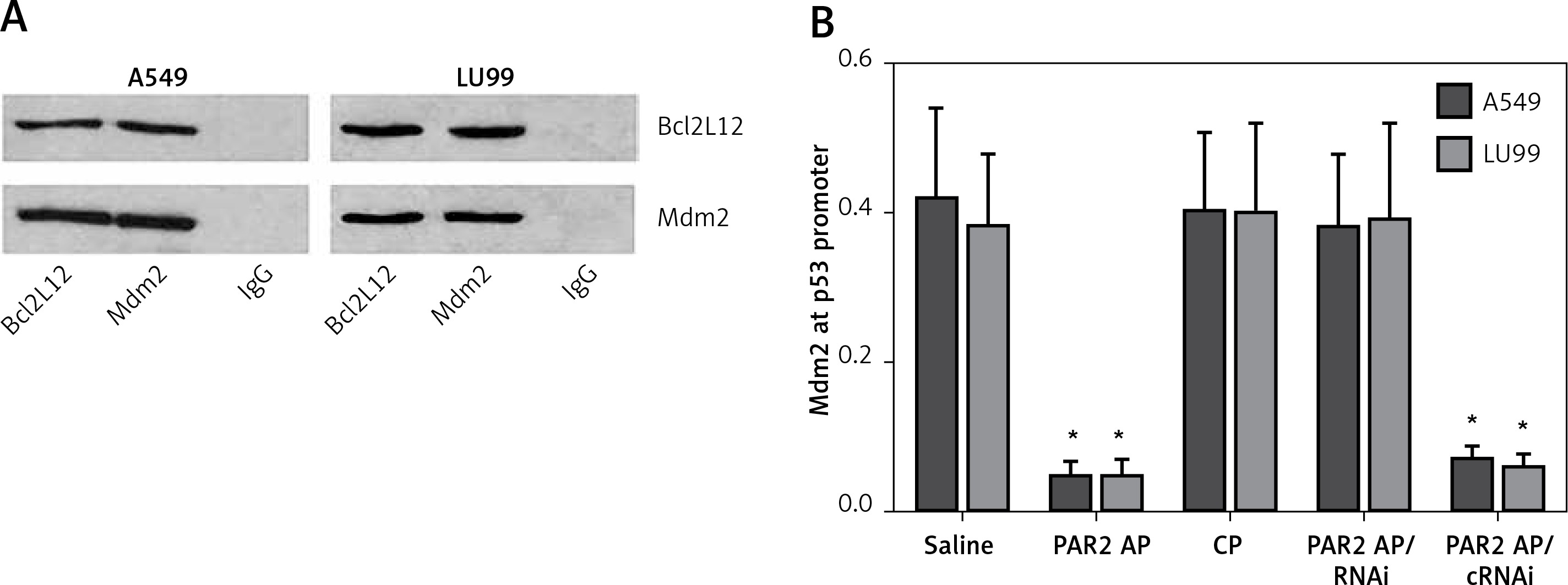
The data reported above imply that the Bcl2L12 interferes with p53 expression in lung cancer cells. To test this, we prepared nuclear extracts from the lung cancer cells after exposure to PAR2 AP for 48 h. The nuclear extracts were analyzed by immunoprecipitation. The results showed that a complex of Bcl2L12 and Mdm2 was detected in the samples (Figure 3 A). We then analyzed the nuclear extracts with ChIP. The results showed that the binding of Mdm2 to the p53 promoter was observed in the lung cancer cells exposed to saline, which was significantly diminished by exposure to PAR2 AP, but not in those lung cancer cells with the Bcl2L12 gene knocked down (Figure 3 B). The results demonstrate that Bcl2L12 can inhibit the p53 gene transcription via binding the transcription factor Mdm2 in lung cancer cells.
Figure 3
Detection of Bcl2-like protein-12 (Bcl2L12)/Mdm2 complex and Mdm2-binding to the p53 promoter in lung cancer cells. A – Bcl2L12/Mdm2 complex in lung cancer cells. B – Levels (mean ± SD) of Mdm2 at the p53 promoter locus in lung cancer cells after the treatment denoted on the X axis
*p < 0.01, compared with the saline group. The data represent 3 independent experiments, RNAi – lung cancer cells were treated with shRNA of Bcl2L12. cRNAi – lung cancer cells were treated with control shRNA.
Bcl2-like protein-12 mediates proteinase-activated receptor-2-reduced lung cancer cell apoptosis
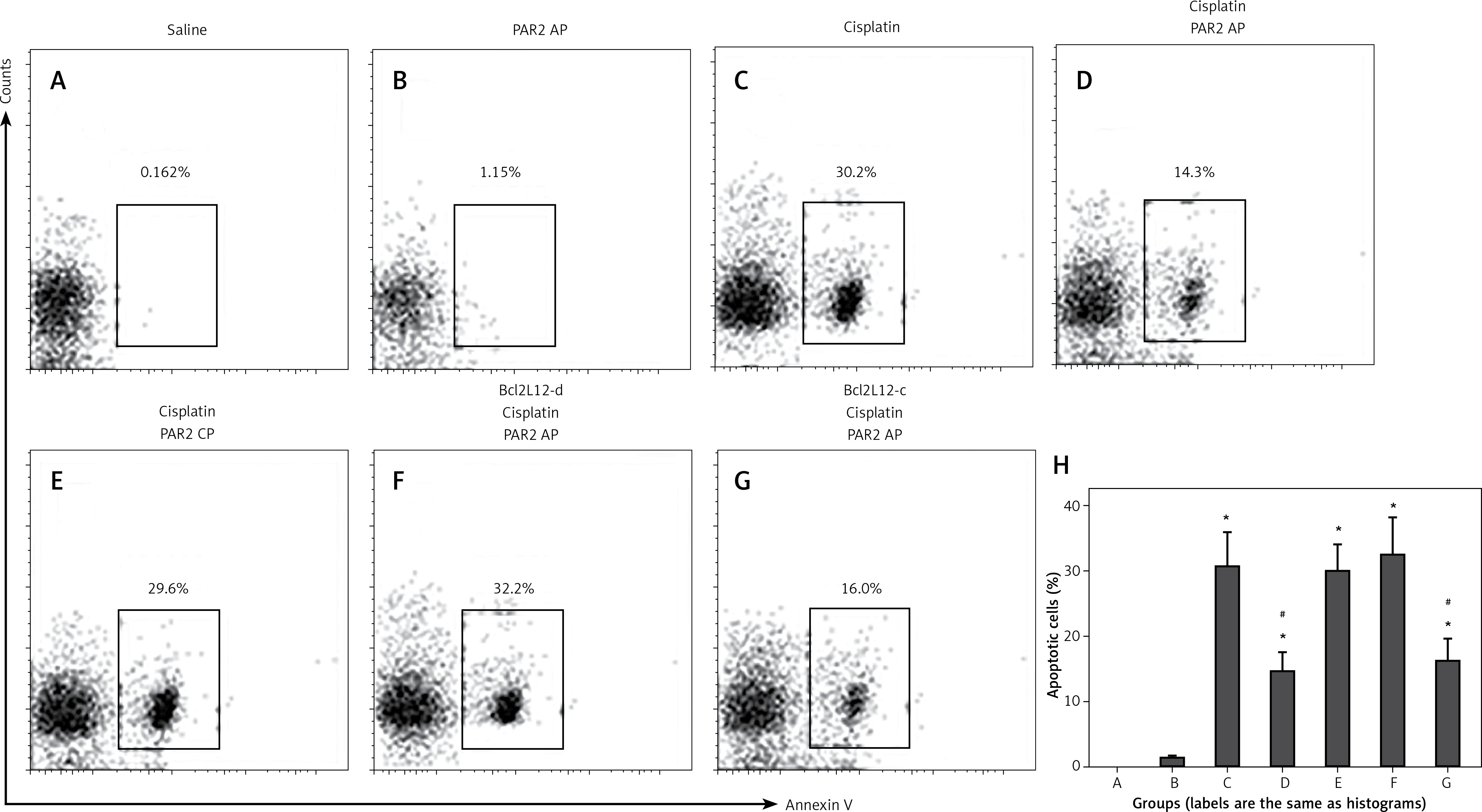
To elucidate whether PAR2 can attenuate apoptosis via inducing Bcl2L12, we treated Bcl2L12-deficient and wild type lung cancer cells with PAR2 AP and/or cisplatin (an apoptosis inducer). The results showed that exposure to saline or PAR2 AP alone did not induce apparent apoptosis in lung cancer cells. Exposure to cisplatin markedly induced apoptosis in the wild type lung cancer cells, which was attenuated by the presence of both cisplatin and PAR2 AP. The results indicate that PAR2 attenuates the effects of cisplatin on inducing lung cancer cell apoptosis. To elucidate whether inhibitory effects of PAR2 were via inducing Bcl2L12, we treated the Bcl2L12-deficient lung cancer cells with both cisplatin and PAR2 AP. Indeed, the inhibitory effects of PAR2 on the cisplatin-induced lung cancer cell apoptosis were blocked (Figure 4).
Figure 4
Proteinase-activated receptor-2 (PAR2) attenuates apoptosis via inducing Bcl2-like protein-12 (Bcl2L12). A–G – The gated cells are apoptotic lung cancer cells after the treatment as denoted above each subpanel. H – summarized data (mean ± SD) of A–G
Bcl2L12-d – lung cancer cells were treated with shRNA of Bcl2L12, Bcl2L12-c – lung cancer cells were treated with control shRNA. The data represent 3 independent experiments. *p < 0.01, compared with group B. #p < 0.01, compared with group B.
Discussion
The present study has revealed the previously unknown phenomenon that activation of PAR2 increased the expression of Bcl2L12 in lung cancer cells. Bcl2L12 bound to Mdm2 to form a complex. The interaction between Bcl2L12 and Mdm2 prevented Mdm2 from binding to the p53 promoter and prevented the expression of p53. Activation of PAR2 attenuated lung cancer cell apoptosis. The data suggest that Bcl2L12 may be a target in the treatment of lung cancer cells.
Bcl2L12 was first found in association with the pathogenesis of glioma by suppressing p53 expression in glioma cells [5]. Latterly, Bcl2L12 was also found in other cancer cells. Tzovaras et al. found that Bcl2L12 expression was positively correlated with the progression of breast cancer and suggested that Bcl2L12 might be a prognostic biomarker for breast cancer progression [16]. High expression of Bcl2L12 in head and neck squamous cancer and several other cancers was reported [17–20]. Our data have added a novel aspect in the study of Bcl2L12 by showing that Bcl2L12 is also expressed by lung cancer cells.
Several reports indicate that cancer cells express PAR2 [21–23]. Our data are in line with those reports; we also found that the lung cancer cell lines, A549 cells and LU99 cells, expressed PAR2 (data not shown). The data showed a novel aspect of PAR2: upon exposure to PAR2 AP, lung cancer cells expressed high levels of Bcl2L12. Bcl2L12 is an anti-apoptosis protein [24]. Published data indicate that Bcl2L12 can suppress caspase-3, caspase-7 and p53 expression in glioma cells. Our study has revealed that Bcl2L12 can also suppress the expression of p53 in lung cancer cells. The data suggest that the inhibitory effect of Bcl2L12 on p53 expression is not limited to glioma cells, but also occurs in other cancer cells.
We found that activation of PAR2 can increase the expression of Bcl2L12 in lung cancer cells. PAR2 can be activated upon exposure to proteolytic enzymes, such as mast cell-derived tryptase [25], MMP-9 [26], etc. Mast cells are distributed extensively in the body. Many factors can activate mast cells to release mediators. Because mast cells are the major effector cells in the allergic response, re-exposure to specific antigens can prevent mast cells from releasing mediators [27]. Thus, the significance of mast cell activation in the regulation of p53 expression is worth being further investigated.
In conclusion, the present study demonstrates that activation of PAR2 attenuates the expression of p53 in lung cancer cells to prevent cancer cell apoptosis via upregulation of Bcl2L12 expression. The data suggest that Bcl2L12 may be a novel target in the treatment of lung cancer.