Phenylketonuria (PKU) is a rare inborn error of metabolism, also known as phenylalanine hydroxylase deficiency or Følling disease. It is inherited in an autosomal recessive manner. The disease affects an average of 1 in 10,000 newborns in Europe [1, 2]. The deficit of phenylalanine hydroxylase leads to the accumulation of phenylalanine in the body. The untreated disease manifests itself with symptoms including those pertaining to the nervous system. These include intellectual disability, seizures, tremor, cerebellar ataxia, behavioural disturbances, and sensory and motor impairment [1, 3]. Jaulent et al. in 2022 after analysing cases reported in French reference centres for inborn errors of metabolism and cases reported in the literature identified 8 new cases of neurological manifestations in patients with phenylketonuria and 22 similar cases in the literature [3]. Thanks to a neonatal screening, PKU can be diagnosed very early. Hence, it can be treated successfully with a phenylalanine-restricted diet or with a tetrahydrobiopterin (BH4) [3].
We present the case of a 36-year-old male patient admitted to the hospital due to moderate tetraparesis. He had noticed tremor of his right lower limb 8 months before. Four months after that both of his left limbs became weaker. Consequently, the patient started having problems with everyday activities. Upon admission to the Neurology Department, the patient initially denied having any chronic disease. Furthermore, the patient reported that he was bitten by a tick over 2 years before the admission to the hospital and was taking antibiotics at the time. During his stay in the ward he disclosed being diagnosed with PKU during a screening test as a newborn. The patient was not aware of the fact that his symptoms could have been caused by PKU. The patient admitted that he had not been following the low-phenylalanine (low-Phe) diet for approximately 10 years. He also had not been attending appointments at the Metabolic Diseases Clinic due to social problems as a person in the crisis of homelessness.
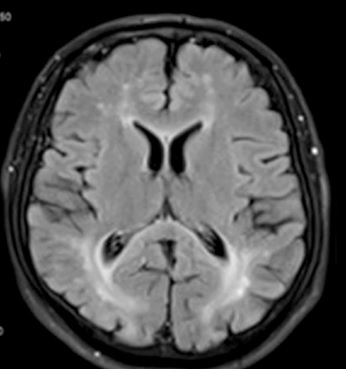
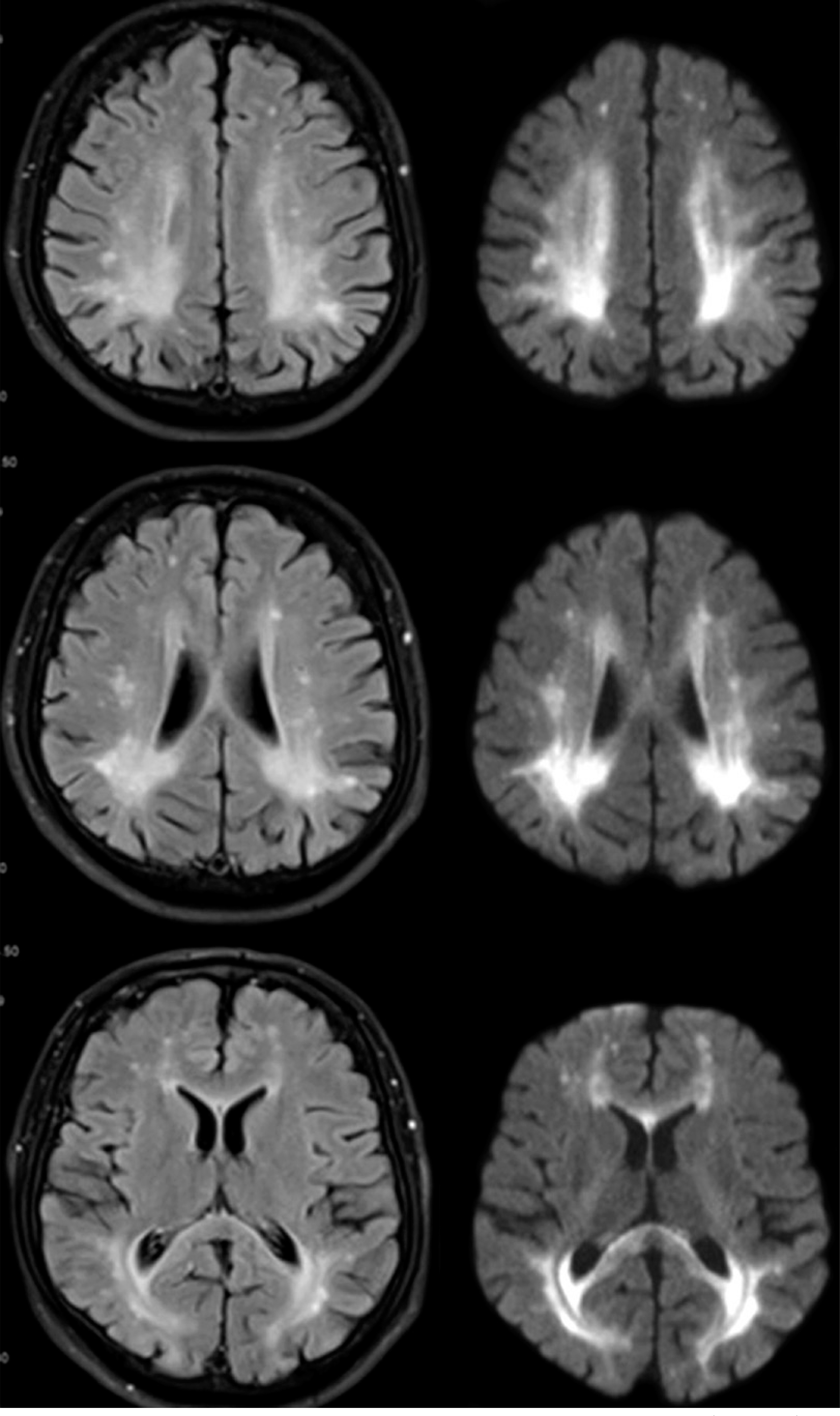
The neurological examination revealed the presence of central paresis of the left facial nerve, moderate spastic tetraparesis (in Lovett scale: 3/5 in both upper limbs, 4/5 in both lower limbs), brisk deep tendon reflexes as well as clonuses and bilateral pathological signs in upper and lower limbs. A brain magnetic resonance imaging (MRI) (Figure 1) found periventricular white matter lesions (FLAIR hyperintensities) with no post-contrast enhancement and reduced diffusion in diffusion-weighted imaging (DWI) in affected regions. The images were reviewed by a neuroradiologist. The brain MRI demonstrated abnormalities consistent with PKU. Electroencephalogram showed only low-voltage activity. A lumbar puncture was performed and laboratory values of the cerebrospinal fluid were within normal range (cytosis – 1 cell/µl, proteins – 34 mg/dl). Laboratory tests revealed a deficiency in folic acid, which was subsequently supplemented. Vitamin B12 levels were within the normal range. Tumor markers, tests for rheumatological diseases, antibodies to Borrelia burgdorferi in the IgM and IgG classes, antibodies to treponema pallidum in the IgM and IgG classes, and HIV test were all negative.
Figure 1
Typical changes for phenylketonuria in brain MRI: periventricular FLAIR hyperintensities (left column) and reduced diffusion in diffusion-weighted imaging (DWI) in affected regions (right column)
Neuropsychological examination found mild cognitive disorders in the field of verbal and visual-spatial fluency and moderate disorders of memory processes. The cognitive impairment corresponded to phenylketonuric encephalopathy.
The patient was consulted by a dietitian and was reminded of principles of the low-Phe diet. A meeting with a social worker was held and options of the further management, including places where a person in the crisis of homelessness can get help and accommodation after discharge, were discussed.
The patient was discharged in good general condition and was referred to the Metabolic Diseases Clinic for further examination.
At the 2-month follow-up, the patient’s neurological condition was stable. A pre-paid low-Phe diet had been previously offered to the patient in the Metabolic Diseases Clinic.
Although phenylketonuria is a rare genetic disorder and newborn screening tests are performed, physicians should be aware of the disease. The vast majority of the people with PKU follow the diet and present no symptoms. However, some adults can manifest signs of the disease, such as individuals with social problems, like the patient described in the case.