Neurofibromatosis type I (NF1) is an autosomal dominant genetic disease caused by a mutation in the neurofibromin 1 gene located on chromosome 17q11.2 [1]. About 50% of patients have a clear family history, while the rest are sporadic or due to radiation [2]. Patients with NF1 also have an increased risk of developing a variety of tumors, including malignant peripheral nerve sheath tumor (MPNST), pheochromocytoma, leukemia, glioma and rhabdomyosarcoma [3]. MPNST is a highly malignant tumor that is closely related to NF1. In NF1 patients, the lifetime risk of MPNST is 8% to 13% [4]. The recovery from NF1-associated MPNST is worse than the sporadic or radiation-related form [5]. Meanwhile, it will also increase the economic and psychological burdens of patients. Therefore, patients with a severe psychological burden should also seek psychological counseling and life help [6].
In this article, we report a case of NF1-associated MPNST in a young man, along with a review of relevant literature.
A year ago, a 26-year-old young man discovered a mass on the proximal lateral side of the right thigh. The mass was significantly enlarged, and its growth accelerated 5 months ago. The patient was hospitalized due to the large tumor and pain. This patient had a family history of NF1. According to the patient’s family, the patient’s mother was diagnosed with multiple malignant tumors of the digestive tract and died as a result. She had NF1 mutations confirmed by sequencing before she died. This time, we also sequenced the patient’s 2 uncles (his mother’s 2 brothers), and through genetic sequencing, we found that the patient’s 2 uncles also have NF1 gene mutations. The patient’s 2 uncles showed a large number of masses on the body surface. His grandfather (mother’s father) had egg-sized lumps on his head when he was young and was diagnosed with NF1, had a surgical resection and no recurrence afterwards. At the time of this study, he was too old to participate (Figure 1). The patient’s physical examination showed scoliosis, multiple cafe-au-lait spots of different sizes all over the body, a huge mass on the lateral side of the right thigh, with medium texture, general mobility and obvious skin veins on the surface of the mass, which had a size of 50 × 33 × 32 cm (Figures 2 A, B). The neurological examination showed no obvious symptoms. Magnetic resonance imaging (MRI) of the right thigh revealed a large soft tissue mass (Figure 3), and therefore a diagnosis of neurofibromatosis was suspected. The bone scan showed a slightly increased area of large flaky radioactive uptake in the right hip and thigh, an uneven distribution of radioactivity, and no abnormalities were observed in the other bones (Figure 4). An incision biopsy suggested neurofibromatosis, and the immunohistochemical staining showed that the mass was positive for vimentin, S-100, Bcl-2, CD99 and weakly positive for CD34. The tumor cells were negative for CKpan, SMA, desmin, NSE, NF and β-catenin and the Ki-67 labeling index was less than 1%. A right lower limb mass resection was performed under general anesthesia. The pathological results showed that round, oval and short fusiform cells proliferated. The cells were abundant, nuclear atypia was obvious, mitosis was common, and there was a massive necrosis in the mass. Immunohistochemical staining showed that the mass was positive for vimentin, β-catenin, Bcl-2, CD99 and there was partial weak positivity for NSE. The tumor cells were negative for CKpan, desmin, S-100, CD34, NF, EMA and SMA and the Ki-67 labeling index was approximately 40% (Figure 5). Whole-exome sequencing was performed with an average sequencing depth of 121.51X in the target region and a capture efficiency of 85.12%. Data analysis revealed two mutations (c.7702C>T, p.Q2568X; c.7765C>T, p.Q2589X) in the NF1 gene, which led to termination codons, resulting in abnormal disruption of protein coding. Notably, we also found a mutation (c.4615T>C, p.S1539P) in CHD5. Combined with the clinical history, a diagnosis of malignant peripheral nerve sheath tumor was made. We advised the patients and their families to receive radiotherapy with 50 Gy in 25 fractions and chemotherapy (liposomal doxorubicin + cisplatin) preoperatively and postoperatively. However, because of the family financial poverty, the patient did not receive neoadjuvant radiotherapy, but actively requested surgical resection. Because the tumor is huge, during the operation we found that the tumor involved the gluteus, the thigh muscle group and sciatic nerve, and deep into the pelvic cavity, the surgical margin still has visible tumor tissue (R2), and the tumor cannot be completely removed. Therefore, we only performed palliative tumor reduction surgery on this patient. After tumor resection, the patient refused further chemoradiotherapy and was discharged. According to our advice, the patient was followed up every month after discharge, and at the 4th month, the patient presented with a recurrence of pain in the operation area and a fist-sized hard mass. The patient was re-hospitalized due to the recurrence of the tumor at the surgical site. Chemoradiotherapy was performed first, and then surgery was considered according to the size of the tumor after chemoradiotherapy. The patient received right thigh mass radiotherapy with 50 Gy in 25 fractions and chemotherapy (liposomal doxorubicin + cisplatin). After chemoradiotherapy, the effect is not satisfactory. We switched to two kinds of tyrosine kinase inhibitors (apatinib + imatinib), but the effect is still not good. However, during the treatment, the patient developed anemia, hypoproteinemia and skin and deep tissue burst into the tumor area. We gave the patient blood transfusion, nutritional support and an infection preventive. The patient refused to undergo the operation and to continue the above treatment. Two months later the patient died of severe malnutrition and infection.
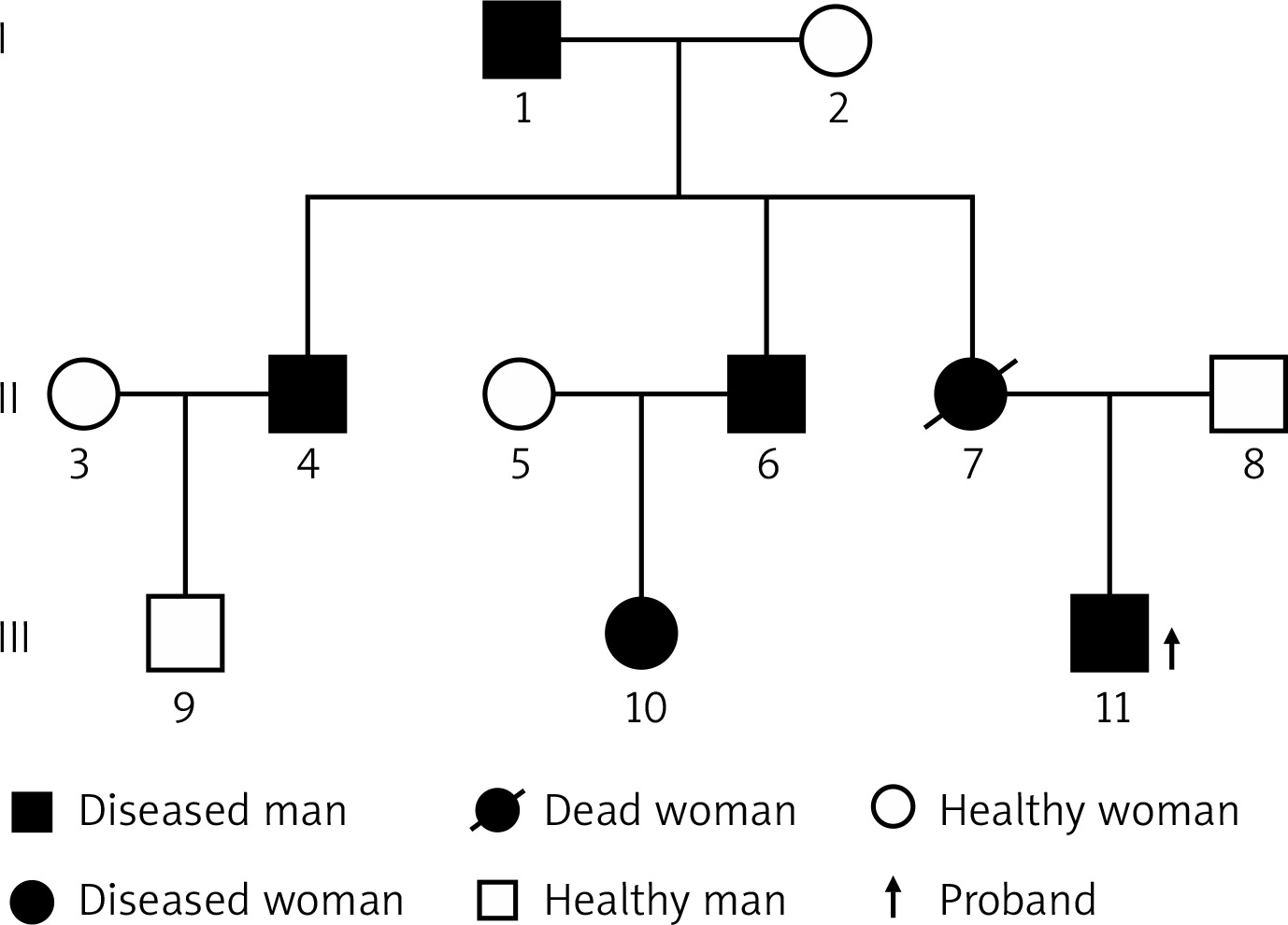
Figure 2
Preoperative photos: A – anterior view, B – norma lateralis, C – norma occipitalis, D – the diameter of the mass reached 95 cm, E – the longitudinal length of the mass reached 50 cm
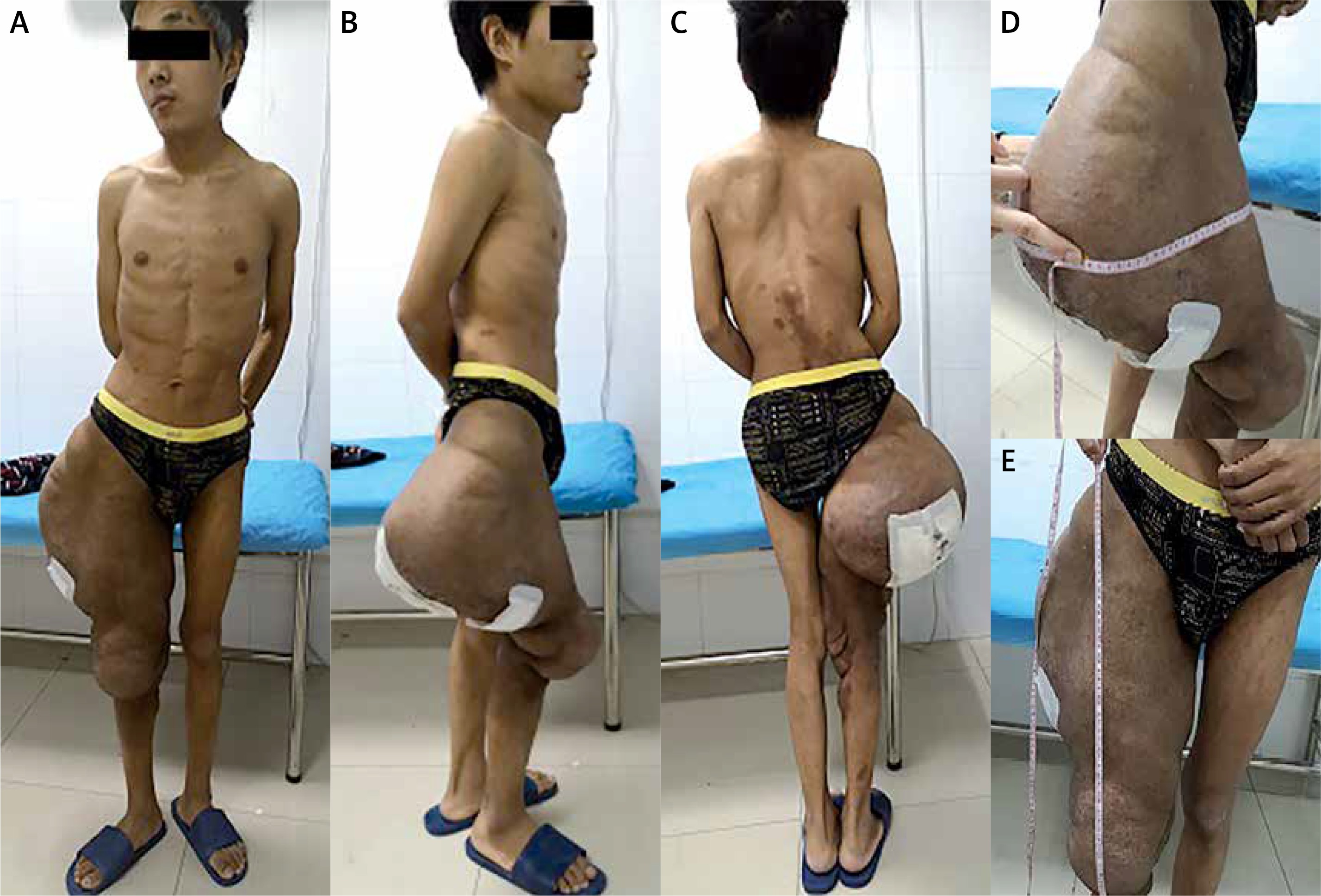
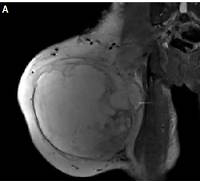
Figure 3
Magnetic resonance imaging showed space occupation in the lateral muscle group and a subcutaneous fat layer in the right thigh. A – Coronal section, B – transverse section
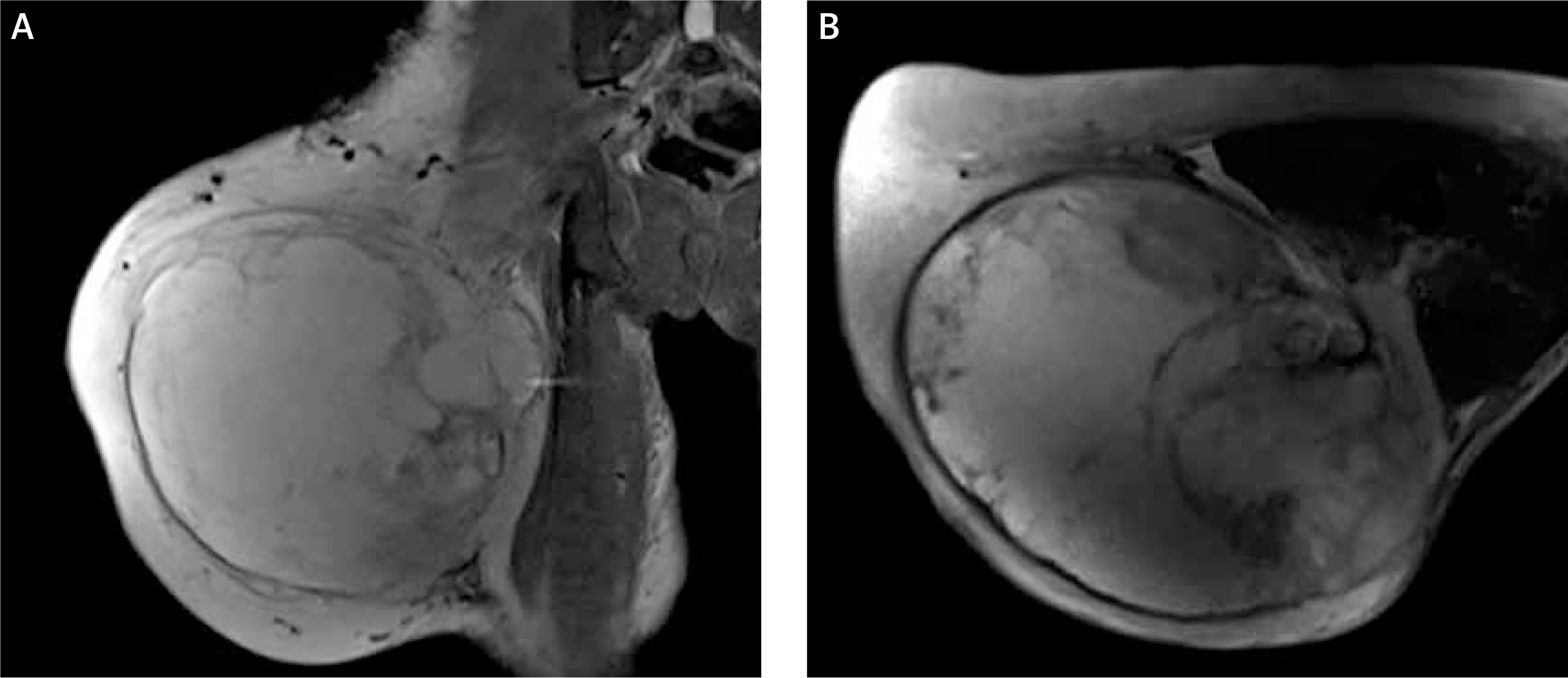
Figure 4
(1) Lateral curvature of the spine;(2) right hip and thigh area-sheet radiodistribution, considering ectopic tumor uptake
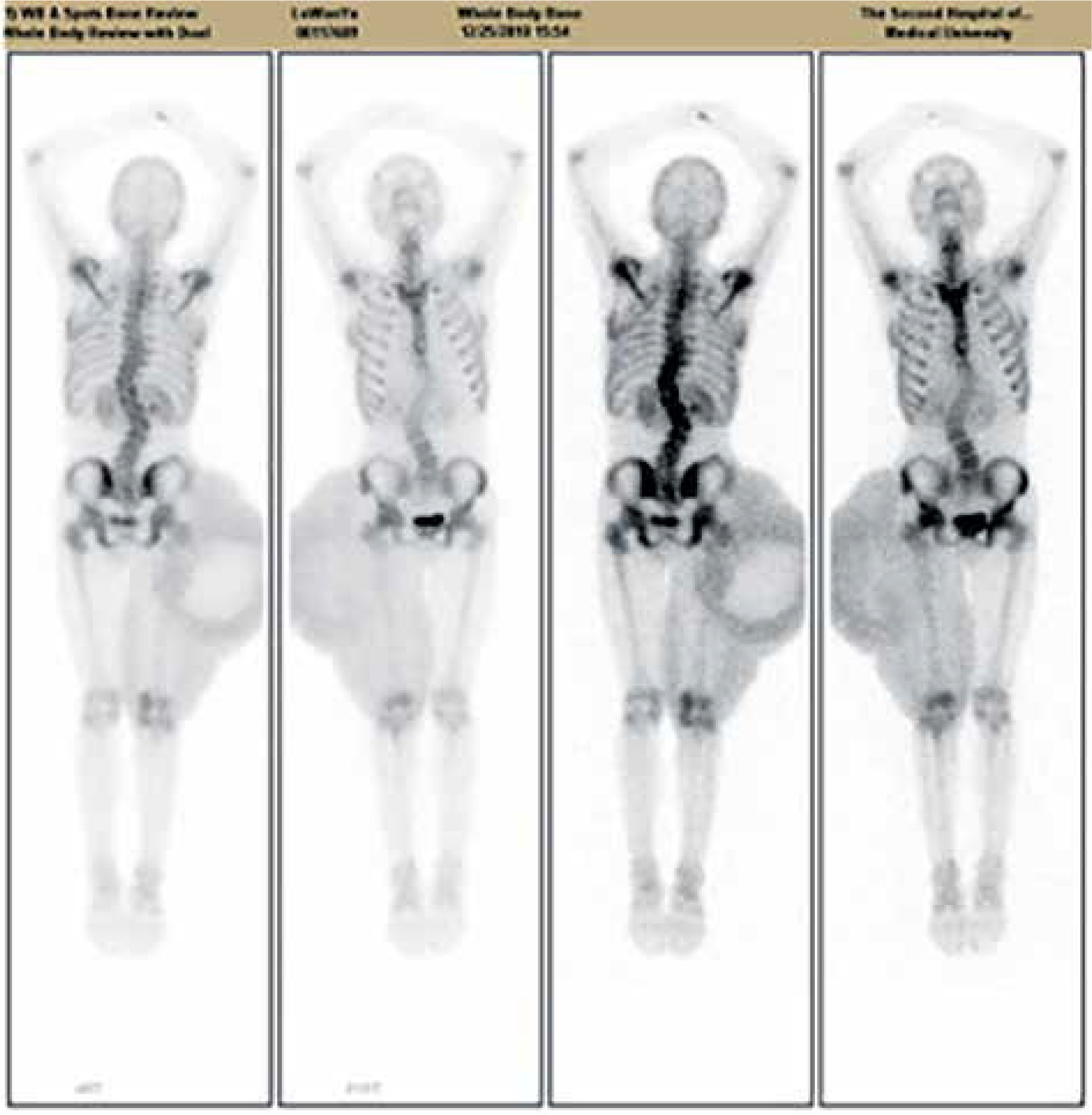
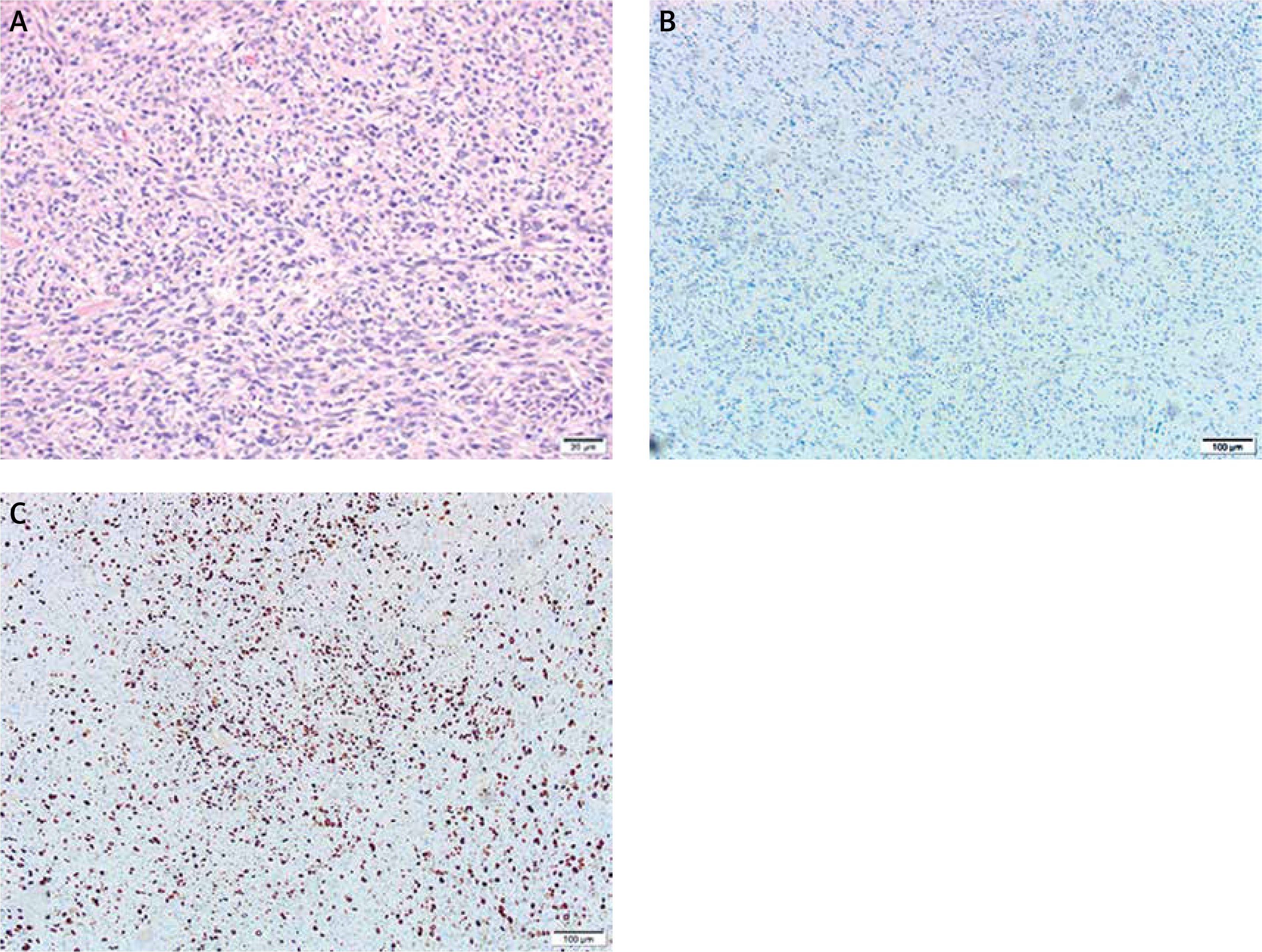
Figure 5
Microscopic findings. A – Under high power microscope, the cells were round, oval, fusiform hyperplasia and obvious heteromorphism, B – immunohistochemistry showed tumor negativity for S-100 protein, C – the Ki-67 labeling index was approximately 40%
NF1 is a cancer predisposition disorder caused by genetic mutations with a general population prevalence of approximatively 1/3000 [1]. Whereas gene mutation data for NF1-associated tumors are even more scarce, whole-exome sequencing, which is becoming popular, will certainly contribute to reducing this scarcity. NF1-associated MPNST is mainly transformed from plexiform neurofibromas (PN), which has high incidence [7]. There was no significant gender difference in incidence and the average age of patients with NF1-associated MPNST was approximatively 10 years earlier compared to non-NF1 [8]. It is most frequently observed in the head and neck, followed by the trunk and limbs, next to the vertebral body [9], intraosseous [10], breast [11], and intestinal [12] cases have also been reported. In this report, the patient’s single mass occurred in the right thigh and its sudden increase indicated the possibility of malignant transformation. The patient’s age also reinforced its possibility as the risk of transformation is higher in late adolescence or youth [13]. The patient in this case was 26 years old and was in a high-risk period of NF1 to MPNST.
MPNST is a subtype of Schwann cell-derived sarcoma, accounting for 5% to 10% of all soft tissue tumors and for a large proportion in NF1 [14], with most of them appearing in PN [15]. Some studies have suggested that atypical neurofibromatosis (ANF) is an intermediate step in the malignant transformation of benign PN to MPNST and that the transition from PN to ANF is mainly driven by CDKN2A/B deletion. Further development from ANF to MPNST may involve extensive chromosomal rearrangements and frequent inactivation of the PRC2 gene [16]. To understand the pathogenesis of neurofibromatosis, we also carried out whole-exome sequencing on the patient’s father and uncle to analyze and compare the faulty genes. Phenotypically, the father is healthy and the uncle is a patient with non-malignant neurofibromatosis. However, according to the whole exome sequencing data analysis, the patient did not have the above typical gene mutation. In this reported case, when we compared the patient’s genes with those of the father, the patient had a mutation (c.4615T>C>p.S1539P) in the CHD5 gene, in addition to the same NF1 gene mutation found in his uncle (his mother’s brother). CHD5, as a new gene mutation site, may become a new research direction in the pathogenesis of NF1-associated MPNST in the future.
At present, MPNST clinical diagnosis is relatively difficult. It is mainly based on the patient’s clinical manifestations, imaging examination, histopathology and immunohistochemistry. NF1 malignant transformation is predominantly manifested by uncontrollable pain or the rapid increase of the mass and the texture from soft to hard. Before admission, the patient had a typical pain in the affected area, a large and fast-growing mass with a relatively hard texture. MRI is the mainstream method of radiological examination, and some authors have also proposed some features that are helpful for MPNST diagnosis, such as a diameter of more than 5 cm, clear boundaries, edema around the tumor, tumor intensity, central necrosis and calcification [17]. The right lower limb of the patient was massive and the center was showing necrosis, which confirmed the MRI results. Although MRI is helpful in determining the tumor location and extent, it cannot distinguish between benign and malignant tumors. Hematoxylin and eosin (H&E) staining of the pathological tissue is usually characterized by fusiform, long-elliptic, easy-to-see nuclear division, pathological nuclear division and map necrosis. The pathological results showed that the histocytes were proliferative, round, oval and fusiform cells with a significant malignant potential. Marker proteins expressed by MPNST included S-100 protein, CD57 and myelinated basic protein with positive expression rates of 50–70%, 50% and 40%, respectively. However, the higher the degree of MPNST malignancy, the lower is the expression rate of S-100. For this case, where the tumor cell S-100 protein was negative, the clinical diagnosis was relatively difficult. The tumor Ki-67 positive rate in this patient was approximatively 40%. The higher the positive rate, the faster the cell division is and the higher is the probability and degree of malignancy. According to the American Joint Committee on Cancer Staging System for Soft Tissue Sarcomas, the patient’s right lower sarcomas were stage III B [18].
Whole-exome sequencing can specifically obtain sequences of protein coding regions and screen out all mutant genes, and is often used in the diagnosis of disease-related genetic characteristics [19]. In addition to the NF1 mutation, we also found mutations in the CHD5 gene using exome sequencing. CHD5 is a tumor suppressor gene that encodes a protein with chromatin remodeling, helicase and DNA binding motifs that are preferentially expressed in nerve and testicular tissues [20]. However, it remains unclear how CHD5 remodels the chromatin structure. At present, there are relatively few reports on how CHD5 inhibits tumor growth. Among them, it is reported that CHD5 can inhibit cell proliferation, induce apoptosis and cell cycle arrest by inhibiting a series of signaling pathways initiated by WEE1 [21]. WEE1 has been recognized as a potential target for tumor therapy. CHD5 is associated with a variety of tumors, such as neuroblastoma [22], breast [23], gastric [24], and liver cancers [25]. CHD5 chromodomains can directly bind to H3K27me3 and participate in neuronal differentiation [26]. CHD5 deletion can hinder neuronal differentiation and lead to the accumulation of undifferentiated precursor cells, a condition that often occurs in aggressive neuroblastoma. Therefore, we infer that CHD5 gene mutation may be an important factor in the development of MPNST in this patient. However, this type of MPNST-related mutation has not been reported in previous literature. In the absence of CHD5, a subgroup of polycomb-repressed genes become abnormally expressed. The mainstream view is that the inactivation of the polycomb repressive complex 2 (PRC2) is caused by inactivating mutations of its two key components, SUZ12 and EED [27], while homozygous PRC2 inactivation induces complete loss of dimethylation at lysine 27 of histone 3 (H3K27me2) and trimethylation at lysine 27 of histone 3 (H3K27me3) [28]. Since some tumors (e.g. melanoma) lose H3K27me3 through mechanisms that are different from PRC2 loss, the loss of H3K27me3 is not significantly specific to these tumors that need to be differentiated. Further studies have confirmed that H3K27me2 loss, also catalyzed by PRC2, is limited to MPNST and is highly consistent with H3K27me3 loss in several tumors that require differentiation. Thus, H3K27me2 loss is more specific for MPNST diagnosis [29]. Whole-exome sequencing showed no mutations or deletions in SUZ12 and EED genes. Therefore we infer that this case of NF1-associated MPNST may be caused by CHD5 mutations.
So far, no single or combined drug treatment can cure MPNST and a complete surgical resection remains the only treatment option for MPNST patients [30]. The success rate of surgical resection of the low risk tumor located at the end of the limb is greatly increased [31]. However, in patients with high-risk soft tissue sarcoma of the trunk and limbs, neoadjuvant therapy is associated with recurrence-free survival and overall survival when the tumor is ≥ 10 cm [32]. Neoadjuvant (preoperative) and adjuvant (postoperative) therapies combined with radiotherapy and chemotherapy are necessary for the treatment of this aggressive tumor. In general, preoperative radiotherapy and chemotherapy can reduce the tumor size, increase the success rate of surgical resection, kill distant metastatic tumor cells, and reduce the risk of tumor metastasis. The mTORC1 inhibitor rapamycin and drugs that enhance oxidative stress can reduce MPNST [33]. Studies and cases have shown that doxorubicin and ifosfamide could significantly reduce the non-surgically resected MPNST characterized by of H3K27me3 loss of expression or extensive metastatic tumors [34, 35]. However, because of the family financial poverty, the patient did not receive preoperative radiotherapy and chemotherapy, but actively requested surgical resection. The risk of recurrence is extremely high because the tumor cannot be completely removed during the operation (R2 excision). We strongly recommend that patients receive postoperative radiotherapy with 50 Gy in 25 fractions and chemotherapy (liposomal doxorubicin + cisplatin). However, the patient and the family refused again. Postoperative radiotherapy and chemotherapy were performed 4 months after the operation due to the obvious mass recurrence at the surgical site. After receiving radiotherapy and chemotherapy, the tumor did not shrink significantly, and the pain remained apparent. In the course of treatment, we should also pay attention to the effect of tumor rupture on the patient’s mental aspects, and often give patients psychological comfort [36]. At the same time, patients also need to know some general knowledge of self-care after tumor rupture [37]. For this malignancy, the postoperative 5-year overall survival rate remains low. It was reported that about 40% to 65% of MPNST cause local recurrence after surgery and about 30% to 60% of malignant peripheric metastases, most commonly lung metastases, result in MPNST-related death in NF1 patients [38]. Radiotherapy can only delay recurrence, but it does not change the survival rate as MPNST is highly resistant to radiotherapy and chemotherapy. Therefore, surgical resection is still the main treatment for patients. Although there is still a lack of an effective molecular targeted therapy, this therapy still has a good prospect [39]. Considering the excessive activation of RAS in RAS-dependent cancers and the further activation of the receptor tyrosine kinase (RTK) pathway to promote cancer development, the authors supposed that RAS-mediated MPNST could be inhibited by targeting one or more RTK(s). Based on experience with other tumors, we could consider the possibility that multiple RTKs are co-activated in MPNST and are involved in several tumors at the same time [40, 41]. It is possible that different MPNST subtypes depend on different RTKs for growth. Therefore, during the treatment, we boldly tried to combine multiple small doses of TKIs to control the development of MPNST of unknown subtypes, while mitigating the side effects caused by a high-dose use of a single TKI. This method alleviates the side effects of the patients, but the inhibitory effect on the tumor is not obvious. Since there is only one case, there may be particularity. We will verify the effectiveness of this method in shrinking tumors through further study.
In conclusion, NF1-associated MPNST is a highly malignant tumor, and the effect of single surgery or medication is not ideal. At present, it is mainly through surgical resection combined with preoperative and postoperative radiotherapy and chemotherapy. However, there are no reports of a complete cure or better results. Molecular targeted therapy can be a good research direction, and many laboratories have also carried out active research in this direction. When other treatments are ineffective, we can try the combination of multiple TKIs to deal with different cell subtypes of tumors and accumulate more case experience. Considering the rapid development of the medical field in recent years and the deepening of basic and clinical research, we believe that in the near future MPNST will also see better treatment methods.