
Introduction
Phenols are a large family of natural and synthetic compounds with known antioxidant activity [1, 2]. A deficit in antioxidant protection and/or excessive production of reactive oxygen species (ROS) in cells causes oxidative stress, which is detrimental to living organisms [3]. Natural phenols are recognized as nutraceuticals, active components of functional food, often used as adjuvants in therapy, or in prevention of different diseases such as cardiovascular diseases [4], dyslipidaemia [5], neurodegenerative diseases [6, 7], and bacterial and viral infections [8–10].
Despite the promising therapeutic and/or preventive effects and high safety profile, their use is limited, mainly because of their poor bioavailability [4]. Therefore, the use of synthesised natural-like derivatives has potential to overcome this limitation.
So far, the most interest has been shown for derivatives of zingerone and curcumin [11, 12]. Zingerone [4-(4-hydroxy-3-methoxyphenyl)-2-butanone] is an active ingredient isolated from dried or heat-treated ginger (Zingiber officinale, family Zingiberaceae). Curcumin [1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione] is an active component of the root of the turmeric plant (Curcuma longa Linn, family Zingiberaceae).
Hydroxylated biphenyls are examples of natural plant-derived polyphenols. Due to their ability to bind to many types of proteins, hydroxylated biphenyls can affect biological processes within living organisms [13, 14]. The most important pharmacophore in their structure consists of two benzene rings bridged by a single covalent bond. The presence of steric hindrance caused by chemical groups positioned close to the single C-C bond can lead to interactions between biphenyl structures and proteins via selective chiral recognition [1, 15, 16]. A C2-symmetry axis in the structure makes the two aromatic rings indistinguishable. This axis facilitates the synthesis of compounds and their interactions with proteins. Due to their specific structure, hydroxylated biphenyls can reduce oxidative stress to a greater extent than both their corresponding natural and synthetic monophenols [17]. The antioxidative activity of hydroxylated biphenyls increases when the phenol hydroxyl groups are located in ortho position to the single C-C bond between the two aromatic rings as it influences intramolecular hydrogen bonding and stabilisation of the generated phenoxyl radical [1, 18]. The presence of a methoxyl group in ortho position to the phenolic hydroxyl group (a guaiacyl unit) and an α,β-unsaturated chain in the 4-position provides even better stabilisation of the generated phenoxyl radical [17, 18].
Phenols, in addition to being antioxidants, can also exhibit pro-oxidant characteristics. Under normal conditions phenoxyl radicals formed during an antioxidative reaction are not pro-oxidative due to their rapid conversion back to non-radicals via polymerisation, enzymatic or non-enzymatic radical reduction reactions. However, phenoxyl radicals can exhibit cytotoxic pro-oxidative activity in the case of free radical life prolongation [19].
As phenols form the core in the structure of numerous drug molecules, a diverse group of phenolic compounds, including natural and natural-like monomers and their C2-symmetric dimers (hydroxylated biphenyls), was formed, and their effect on oxidative stress was investigated in this study. The antioxidative capability of selected monomers and their C2 symmetric dimers was determined using a number of in vitro assays. Moreover, the way different phenolic structures behave in a biological matrix (blood serum) in regards to their interactions, conformational changes and formation of hydrogen bonds was analysed. Many of the compounds were synthesized for the first time ever.
The goal of this study was to identify those representatives whose pharmacophores have the strongest antioxidant and the lowest prooxidant activity in in vitro conditions and thus determine which phenolic pharmacophore is the most promising for potential further drug development, comparing its activities with Trolox, a hydrosoluble vitamin E analogue as a proven antioxidant substance [20].
Material and methods
Reagents and solvents were of analytical reagent grade and bought from Aldrich Chemie (Steinheim, Germany) and Merck (Darmstadt, Germany).
Compounds 3 and 32 were purchased from Chemos GmbH (Regenstauf, Germany), compounds 22 and 24 were bought from Sigma-Aldrich (Milan, Italy) and used without purification. Compounds 4 and 6-9 were prepared according to procedures described by Marchiani et al. [21], compound 10 as described by Cook et al. [22], compound 12 as described by Choi et al. [23], compound 16 as described by Tatsuzaki et al. [24], compound 18 as described by Varro et al. [25], compounds 20, 21 and 41 as described by Dettori et al. [26], compound 42 as described by Oufensou et al. [27], compound 33 as described by Lin et al. [28], compound 34 as described by Kong et al. [29], and compounds 35 and 36 as described by Maioli et al. [15].
The purity of all new compounds was judged to be > 98% by 1H NMR and 13C NMR spectral determination.
Lipophilicity of compounds 1-36 was estimated by ChemBioDraw Ultra 13.0 software (Cambridge Soft) using the logarithm of the partition coefficient for n-octanol/water (logP) and listed in Table I.
Table I
Structural formulas of tested compounds and Oxy Score values
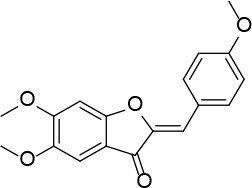
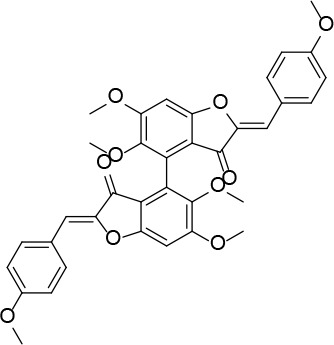
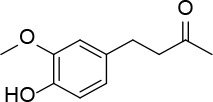
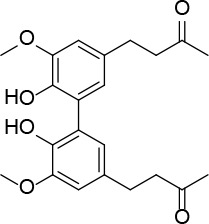
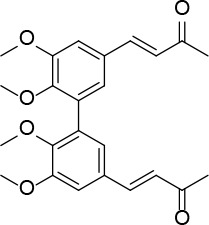
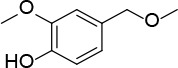
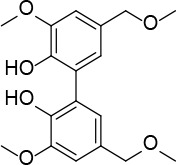
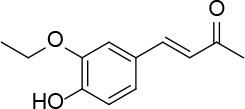
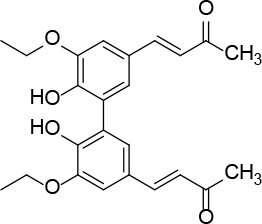
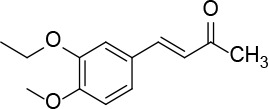
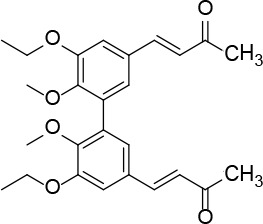
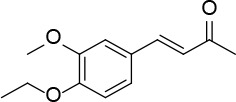
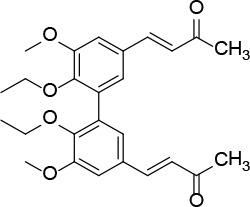
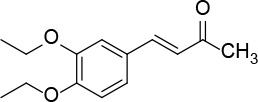
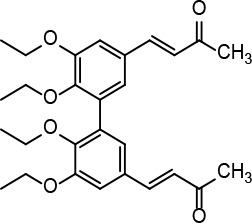
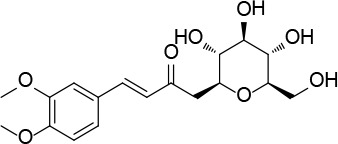
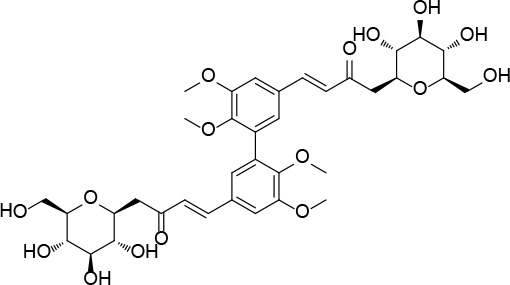
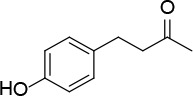
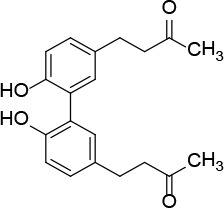
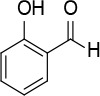
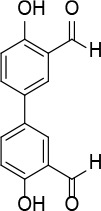
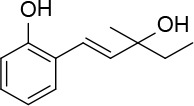
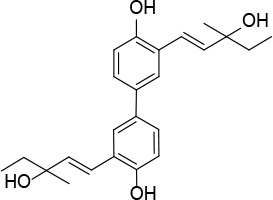
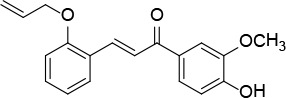
[i] *Aurone derivatives: compounds 1 (monomer) and 2 (dimer); zingerone derivatives: compounds 3 (zingerone, monomer), 4 (zingerone, dimer) and 5 (dimer); dehydrozingerone derivatives: compounds 6 (dehydrozingerone, monomer), 7 (dehydrozingerone, dimer), 8 (monomer) and 9 (dimer), 16 (OEt-dehydrozingerone, monomer), 17 (OEt-dehydrozingerone, dimer); vanillyl alcohol methyl esters: compounds 10 (monomer) and 11 (dimer); ethyl vanillin 3-buten-2-one derivatives: compounds 12, 14 and 18 (monomers); 13,15 and 19 (dimers); glucosylated dehydrozingerone compounds: compounds 20 (monomer) and 21 (dimer); 4-(3-hydroxybutil-3-on) phenol (raspberry ketone): compounds 22 (raspberry ketone, monomer) and 23 (raspberry ketone, dimer); salicylaldehyde and its 4,4’-dihydroxybiphenyl derivative: compounds 24 (monomer) and 25 (dimer); prenylated phenol and 4,4’-dihydroxybiphenyl derivative: compounds 26 (monomer) and 27 (dimer); chalcones and 4,4’-dihydroxybiphenyl chalcones: compounds 28 and 30 (monomers); 29 and 31 (dimers); magnolol: compound 32 (magnolol), and methylmagnolol derivatives: compounds 33 and 34; monoacetylmagnolol: compound 35, and diacetylmagnolol: compound 36. **The solubility of all compounds was 15 mg/ml in DMSO (dimethyl sulfoxide), and concentrations of 10 mg/ml DMSO were used in this study for all compounds tested.
Analysis of antioxidant activity was performed using a microplate reader (BioTek, Winooski, Vermont, USA) and ILAB 300+ automatic analyser (Instrumentation Laboratory, Milan, Italy). Melting points were estimated with a Büchi 530 melting point apparatus in open capillaries and are uncorrected. All 1H NMR and 13C NMR spectra were acquired with a Varian VXR 5000 spectrometer at 399.94 MHz and 75.42 MHz respectively; all spectra were run at room temperature in CDCl3 solution (if not otherwise indicated). Chemical shifts are reported in ppm (δ) on scale downfield from TMS as an internal standard. Signal patterns are indicated as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) or dd (double of doublets). Determination of elemental analyses was done using a Perkin-Elmer elemental analyser, model 240 C. Acetone was distilled from CaCl2. Purification was achieved by silica gel column chromatography using silica gel 60 (230–400 mesh, Kiesgel, EM Reagents) eluting with appropriate solution in the stated v:v proportions. Reaction progress was monitored by thin layer chromatography, 0.25 mm thick pre-coated silica plates (Polygram Sil G/UV254, Macherey-Nagel) and spots were detected under UV light.
Chemical synthesis of new compounds
Neutral alumina (13 g) was added to a solution of 4-methoxybenzaldehyde (0.88 g, 6.46 mmol) and 5,6-dimethoxybenzofuran-3(2H)-one [30] (0.42 g, 2.15 mmol) in dichloromethane (10 ml). The reaction mixture was thoroughly mixed at room temperature for 6 h. The solvent was rotoevaporated, to give the solid product which was purified by crystallization from dichloromethane-petroleum ether to afford 1 (Figure 1) as a yellow solid (0.41 g, 60%): mp 199–200°C (lit54 199-201); 1H NMR δ 3.86 (s, 3H), 3.91 (s, 3H), 4.01 (s, 3H), 6.81 (s, Hz, Ar, 1H), 6.82 (s, 1H), 6.96 (d, J = 8.8 Hz, Ar, 2H), 7.18 (s, Ar, 1H), 7.85 (d, J = 8.8 Hz, Ar, 2H); 13C NMR δ 55.34, 56.31, 56.61, 95.51, 103.90, 112.29, 113.01, 114.37, 125.15, 133.15, 146.47, 146.88, 157.35, 160.78, 163.06, 183.10; Anal. Calcd. for C18H16O5: C, 69.22; H, 5.16; Found: C, 69.33; H, 5.26.
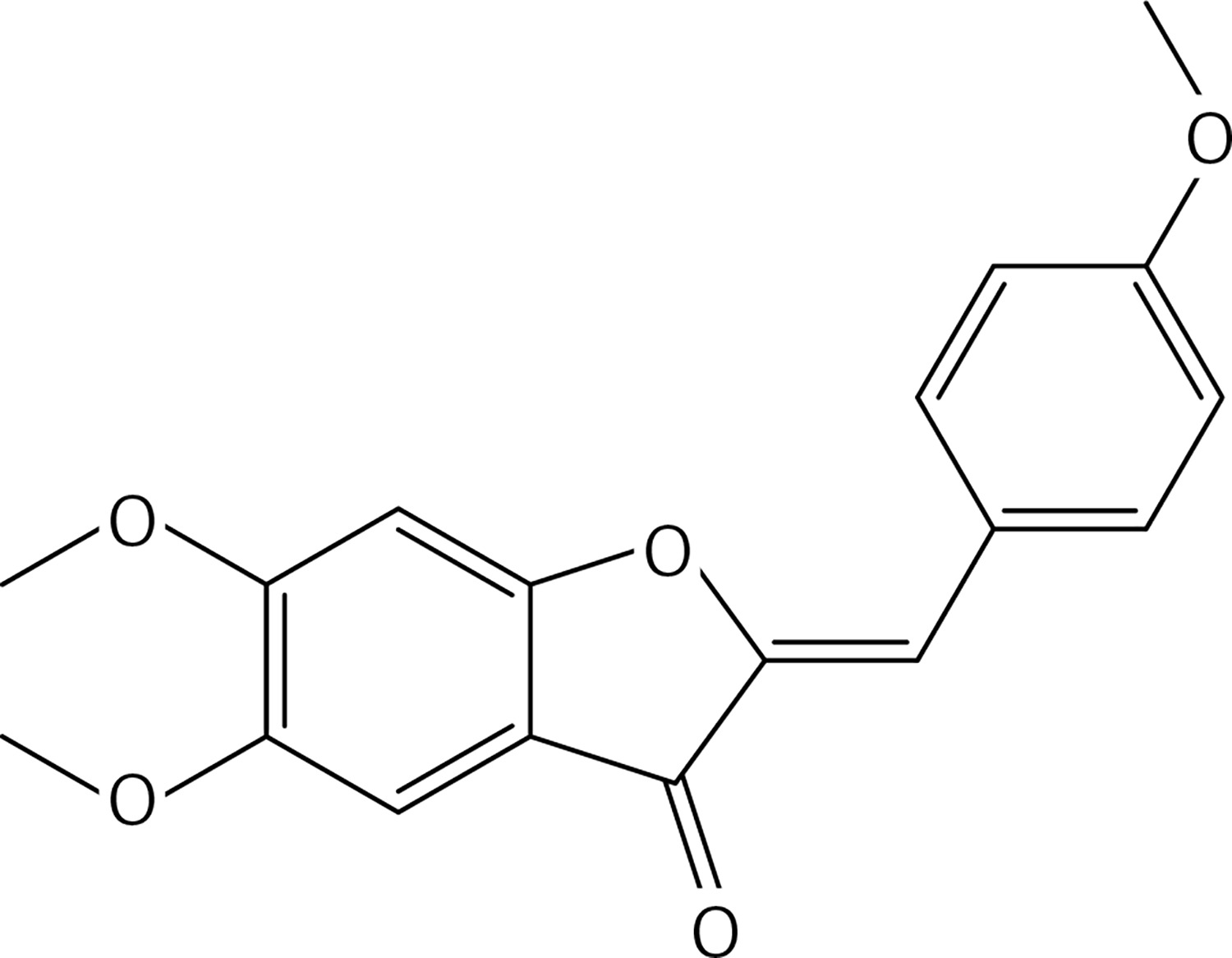
Neutral alumina (2 g) was added to a solution of 4-methoxybenzaldehyde (0.13 g, 1 mmol) and compound 37 (0.13 g, 0.33 mmol) in dichloromethane (10 ml). The reaction mixture was thoroughly mixed at room temperature for 6 h. Removal of the solvent yielded a solid product which was purified by crystallization from dichloromethane-petroleum ether to afford 2 (Figure 2) as a yellow solid (0.12 g, 59%): mp 190–191°C; 1H NMR δ 3.63 (s, 6H), 3.84 (s, 6H), 4.01 (s, 6H), 6.61 (s, 2H), 6.91 (s, Ar, 2H), 6.94 (d, J = 8.4 Hz, Ar, 4H), 7.80 (d, J = 8.4 Hz, Ar, 4H); 13C NMR δ 55.33, 56.35, 61.06, 96.07, 111.61, 112.41, 114.32, 124.97, 125.35, 132.98, 143.51, 146.91, 160.31, 160.59, 164.15, 182.23; Anal. Calcd. for C36H30O10: C, 69.45; H, 4.86; Found: C, 69.39; H, 4.96.
Figure 2
(2Z,2’Z)-5,5’,6,6’-tetramethoxy-2,2’-bis (4-methoxybenzylidene)-[4,4’-bibenzofuran]-3,3’(2H,2’H)-dione (2)
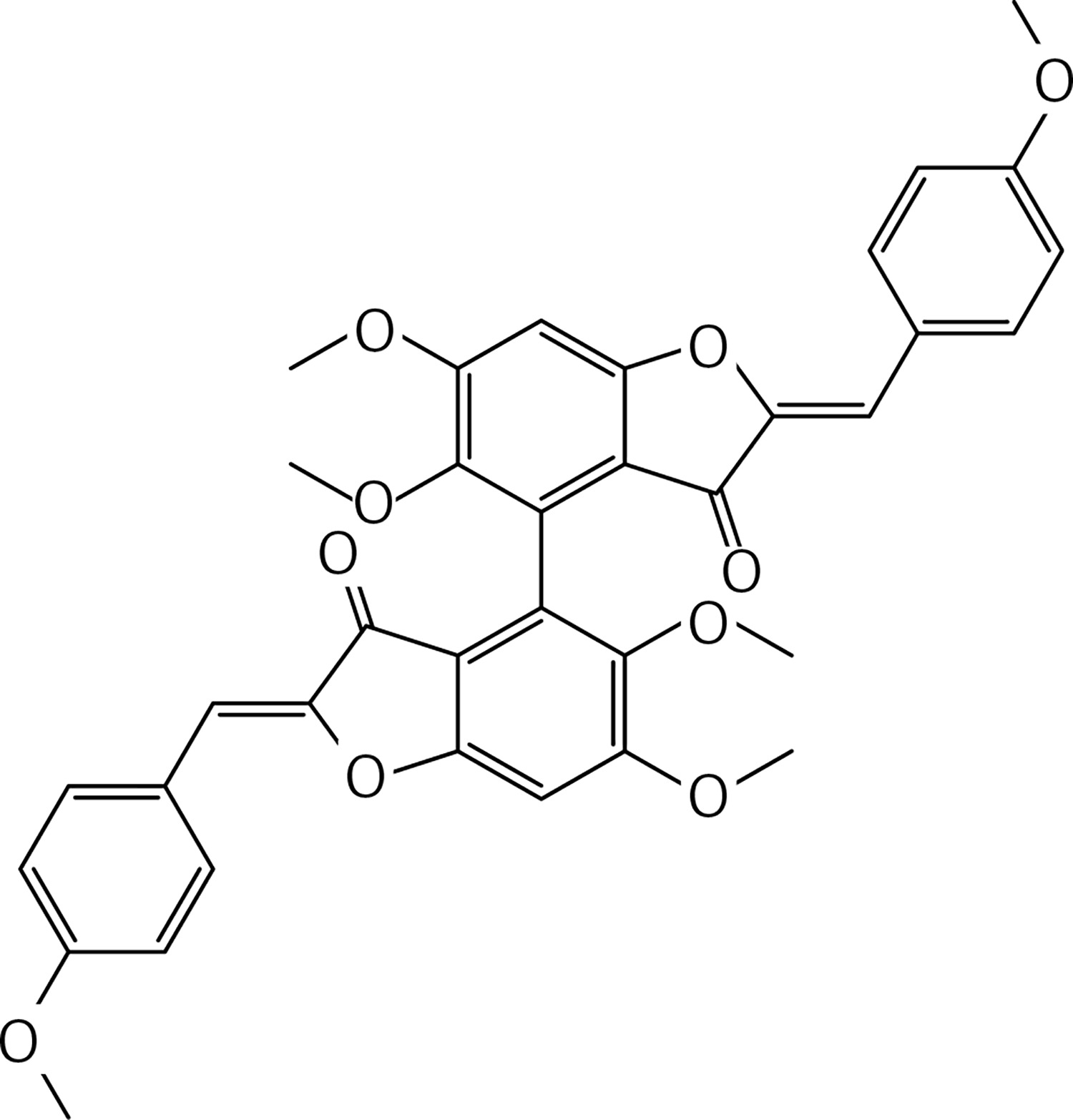
To a solution of compound 38 (0.21 g, 0.89 mmol) in dichloromethane (15 ml), a solution of molybdenum (V) chloride (0.48 g, 1.78 mmol) in dichloromethane (10 ml) was added at 0°C and under N2. The mixture was stirred at 0°C for 45 m. Water was cautiously added. The solution was extracted with dichloromethane and dried over Na2SO4. The crude product was purified by column chromatography using a 1 : 1 mixture of petroleum: acetone as eluent, to give 5 (Figure 3) as a yellow oil (2.16 g, 60%): 1H NMR δ 1.99 (s, 6H), 2.60-2.73 (series of m, 8H), 3.87 (s, 6H), 6.62 (s, Ar, 2H), 6.71 (s, Ar, 2H); 13C NMR δ 27.05, 29.87, 45.12, 55.91, 111.34, 116.18, 130.62, 132.97, 143.38, 145.81, 208.35; Anal. Calcd. for C22H26O6: C, 62.55; H, 5.14; Found: C, 62.54; H, 5.12.
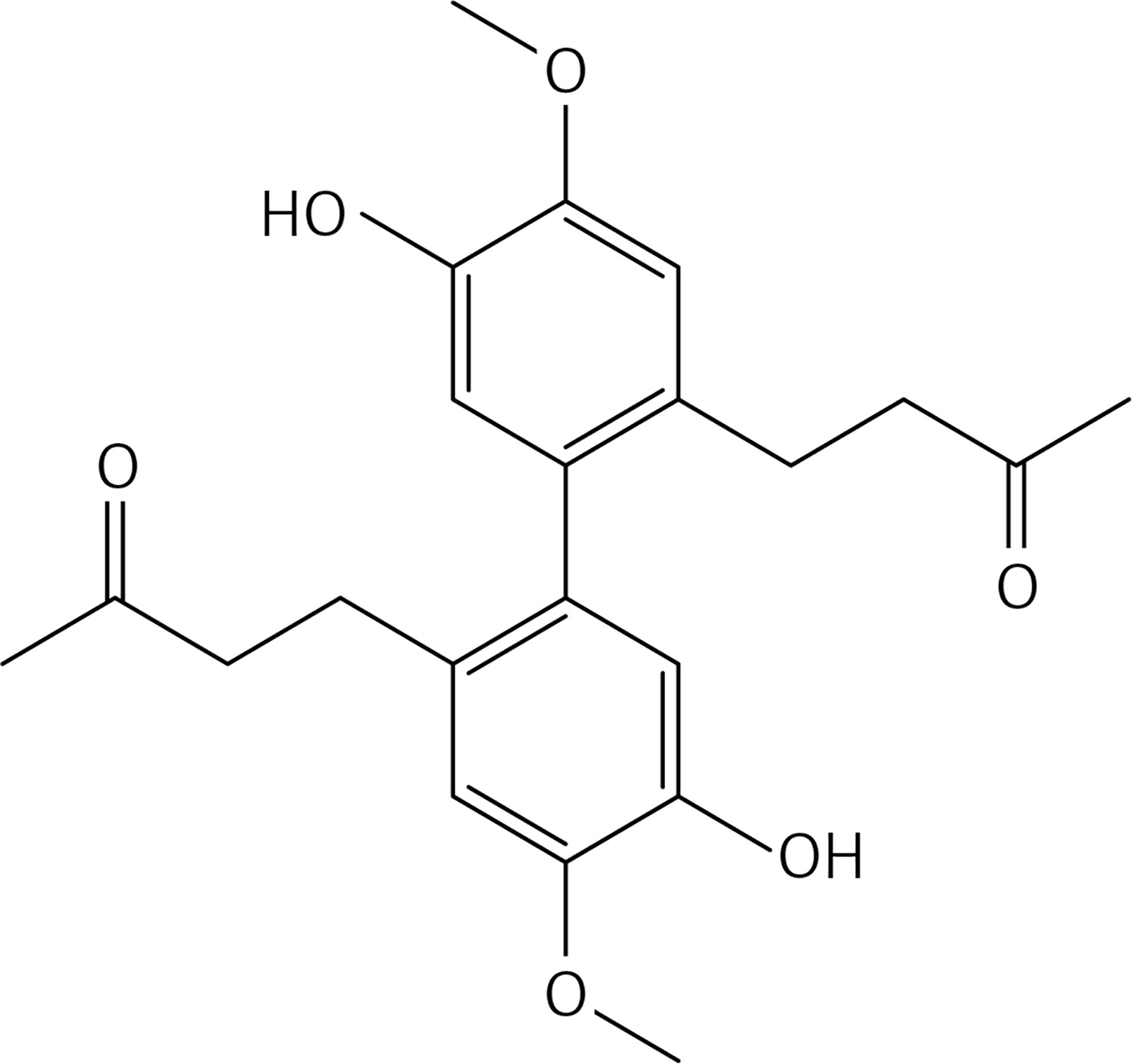
To a solution of 10 (1.6 g, 10.1 mmol) in dry dichloromethane (20 ml), methyl-tributylammonium permanganate (MTBAP) [31] (1.61 g, 5.05 mmol) in dry dichloromethane (15 ml) was added dropwise at room temperature and under N2. The solution was stirred at room temperature for 30 min. An aqueous solution of Na2S2O5 (50 ml) was added. The organic layer was separated, washed with water, dried over Na2SO4 and evaporated to afford 11 (Figure 4) as a white solid. Flash chromatography using a 1 : 2 mixture of ethyl acetate: petroleum ether as eluent gave 11 as a white solid (1.03 g, 61%): mp 103–104°C; 1H NMR δ 3.31 (s, 6H), 3.87 (s, 6H), 5.37 (s, 4H), 6.85 (d, J = 2 Hz, Ar, 2H), 6.94 (d, J = 2 Hz, Ar, 2H); 13C NMR δ 55.51, 56.86, 74.20, 110.96, 122.93, 125.02, 129.39, 143.22, 147.73; Anal. Calcd. for C18H22O6: C, 64.66; H, 6.63; Found: C, 64.76; H, 6.69.
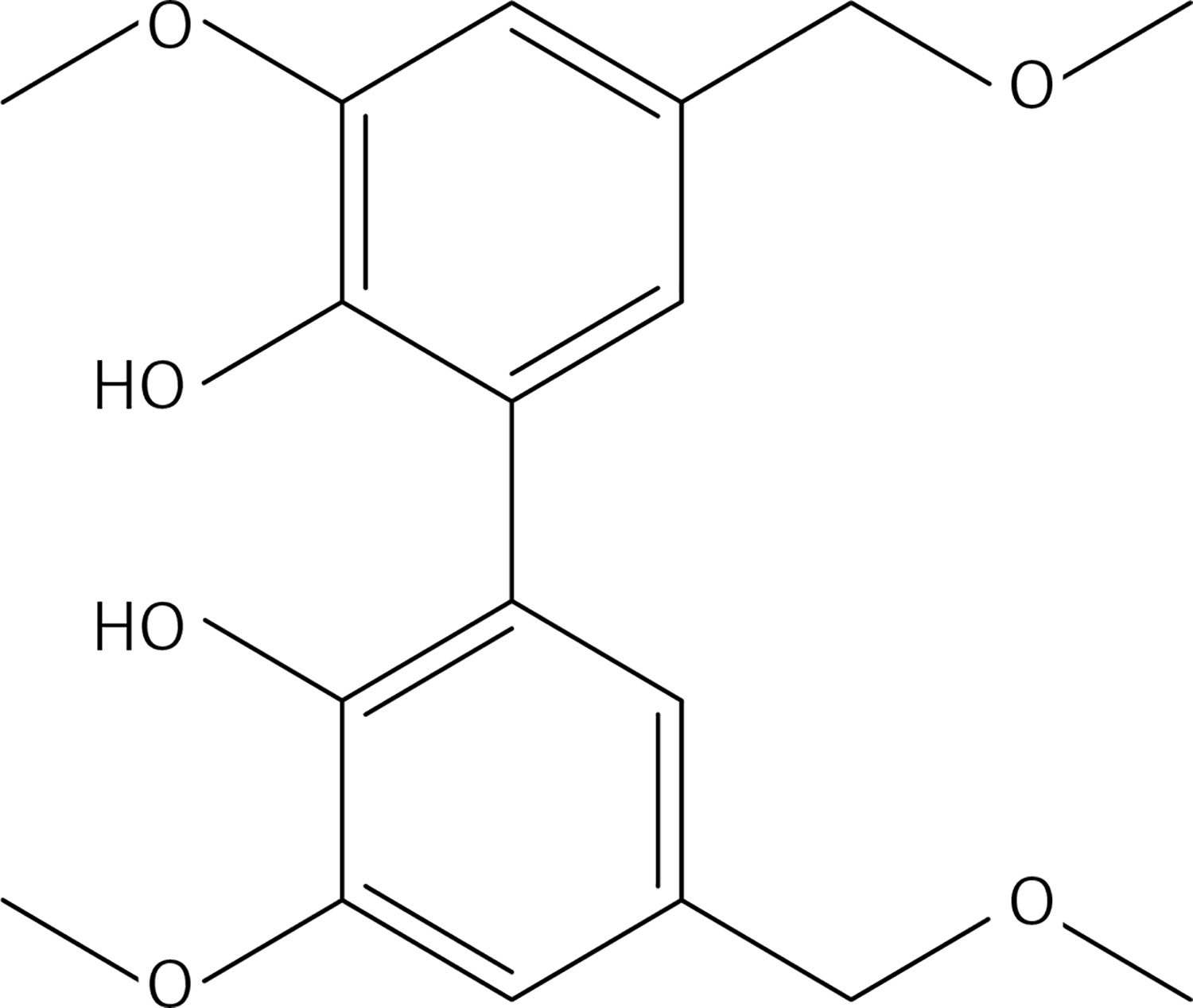
An aqueous solution (1 N) of LiOH (40 ml, 40.0 mmol) was added dropwise to a stirred solution of 39 (2.17 g, 6.6 mmol) in acetone (50 ml) at room temperature and under N2. The mixture was stirred at 60°C for 12 h, 10% HCl and water were cautiously added. The resulting precipitate was filtered, washed with water and dried with Na2SO4. The product was purified by column chromatography using a 1 : 1 mixture of petroleum : ethyl acetate as eluent, to obtain 13 (Figure 5) as a yellow solid (2.16 g, 80%): mp = 100–101°C; 1H NMR δ 1.50 (t, J = 6.4 Hz, 6H), 2.35 (s, 6H), 4.20 (q, J = 6.4 Hz, 4H), 6.20 (bs, 2H), 6.59 (d, J = 16.4 Hz, 2H), 7.01 (d, J = 1.6 Hz, Ar, 2H), 7.17 (d, J = 1.6 Hz, Ar, 2H), 7.47 (d, J = 16.4 Hz, 2H); 13C NMR δ 14.81, 27.32, 56.01, 109.55, 123.56, 125.16, 125.28, 126.40, 143.69, 145.63, 146.55, 198.38; Anal. Calcd. for C24H26O6: C, 70.23; H, 6.38; Found: C, 70.24; H, 6.32.
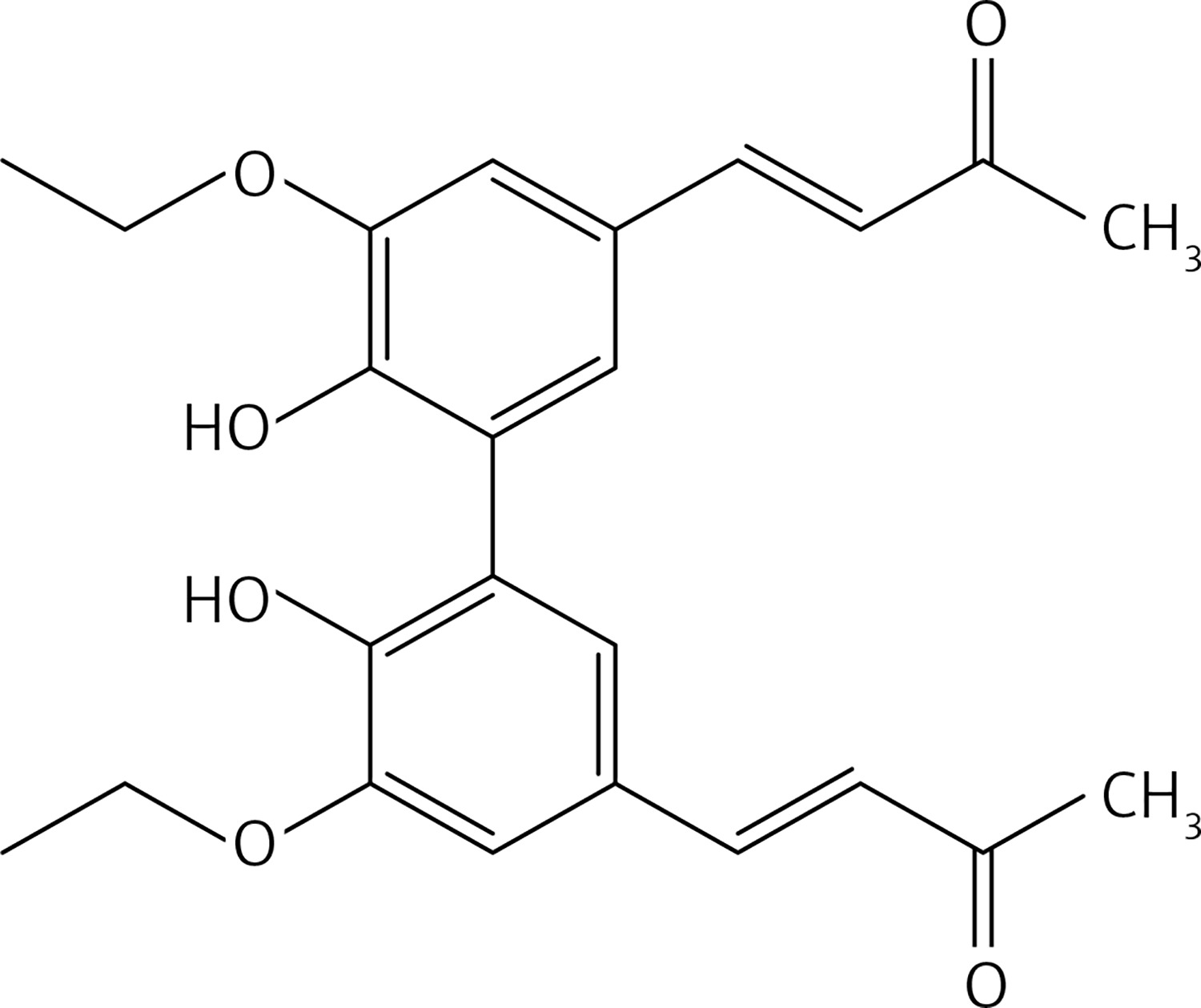
General procedure of phenolic-OH group protection (compounds 14, 15, 17,19, 40)
To a solution of phenol or biphenol (one equivalent) and K2CO3 (1.1 equivalents for monomer, 2.2 for dimers) in dry acetone, alkyl halide (methyl iodide or ethyl bromide or allyl bromide) (1.1 equivalents for monomer, 2.2 for dimers) was added under N2 at room temperature. The reaction mixture was stirred at 56°C for 12 h. Acetone was evaporated and 10% HCl and water were added. The solution was extracted with diethyl ether, dried over Na2SO4 and evaporated. The crude was purified by flash chromatography to obtain the corresponding O-alkylated phenol.
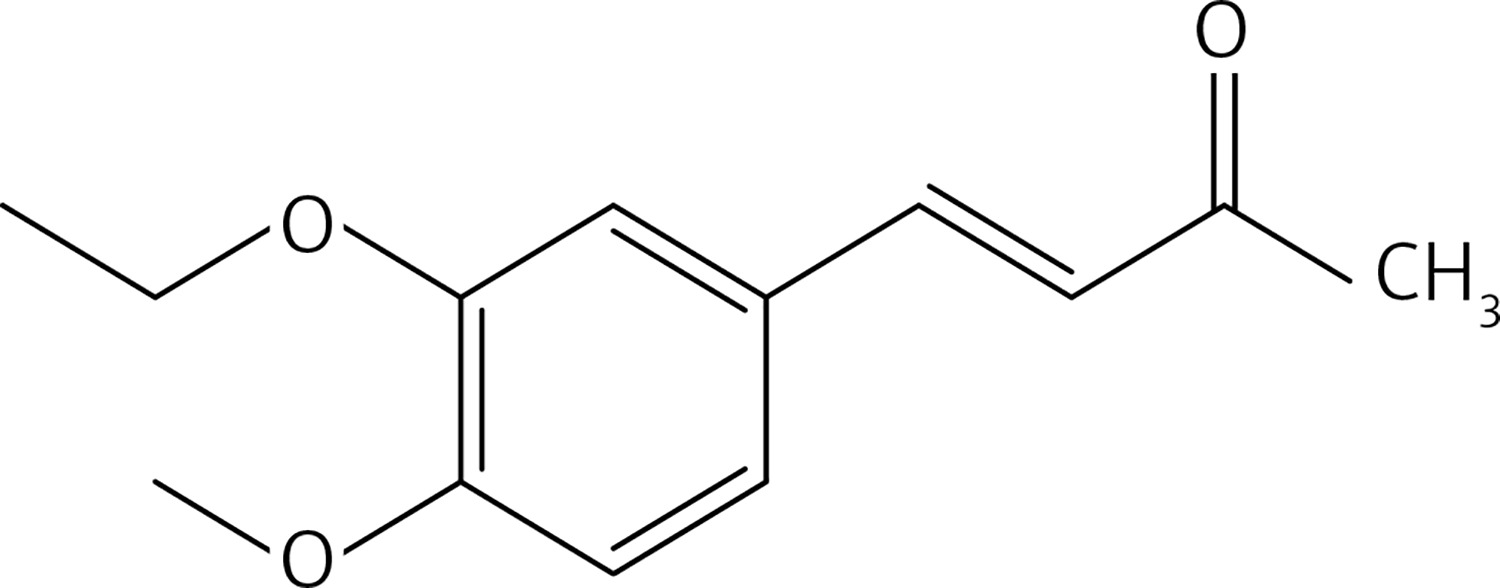
From 12, compound 14 (Figure 6) was obtained after flash chromatography (ethyl acetate : petroleum ether 1 : 5) as a yellow solid (0.42 g, 92%): mp 145–147°C; 1H NMR δ 1.49 (t, J = 6.8 Hz, 3H), 2.35 (s, 3H), 3.92 (s, 3H), 4.13 (q, J = 6.8 Hz, 2H), 6.61 (d, J = 16.4 Hz, 1H), 6.87 (d, J = 8.4 Hz, Ar, 1H), 7.07 (d, J = 2 Hz, Ar, 1H), 7.15 (dd, J = 2, 8.4 Hz, 1H), 7.45 (d, J = 16.4 Hz, 1H); 13C NMR δ 14.72, 27.33, 56.01, 64.36, 110.97, 111.23, 122.91, 125.17, 127.25, 143.66, 148.57, 151.62, 204.49; Anal. Calcd. for C13H16O3: C, 70.89; H, 7.32; Found: C, 70.94; H, 7.36.
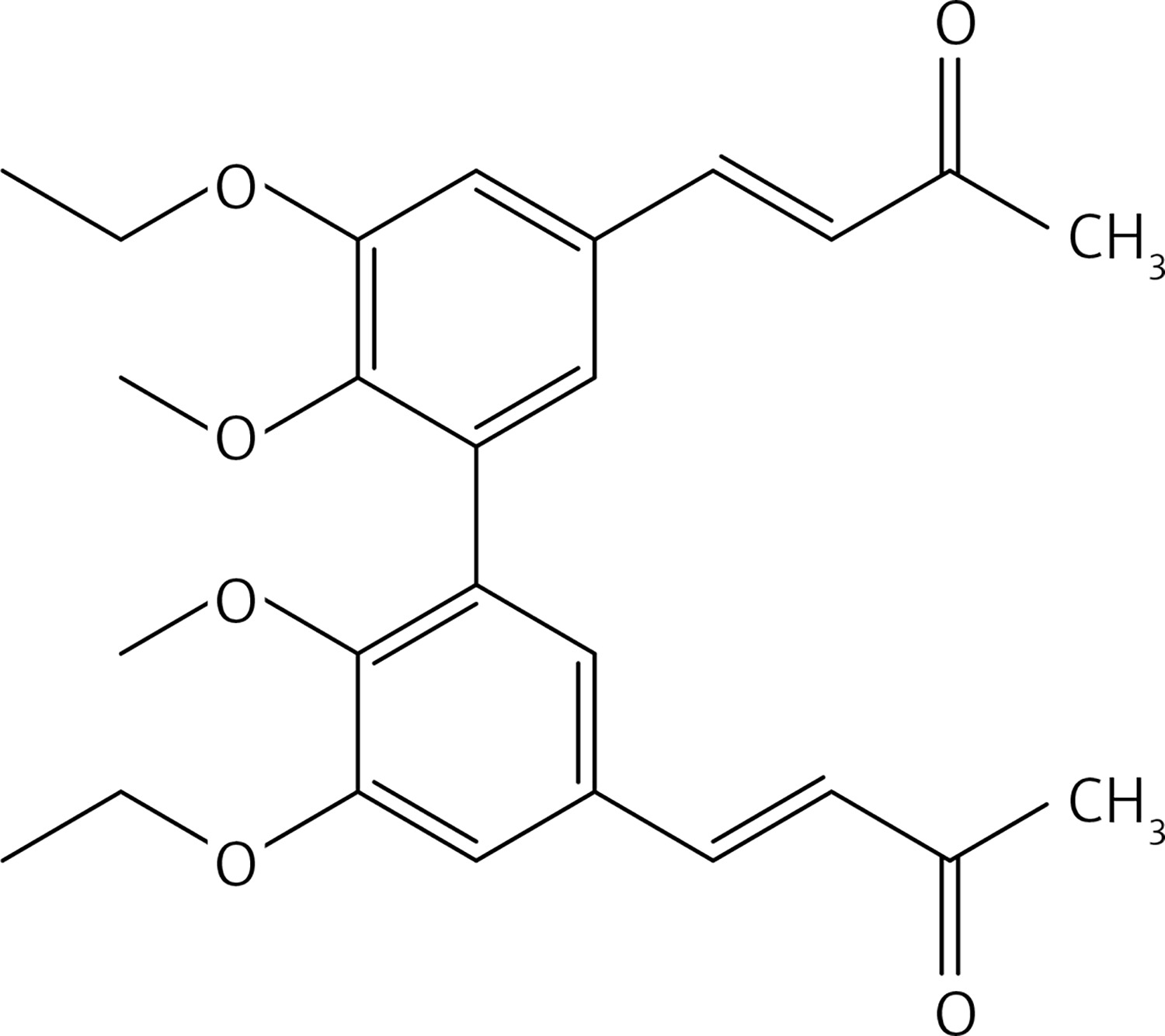
From 13, compound 15 (Figure 7) was obtained after flash chromatography (ethyl acetate : petroleum ether 1 : 1) as a yellow solid (0.84 g, 91%): mp 141–142°C; 1H NMR δ 1.48 (t, J = 7.2 Hz, 6H), 2.34 (s, 6H), 3.72 (s, 6H), 4.13 (q, J = 7.2 Hz, 4H), 6.62 (d, J = 16.4 Hz, 2H), 7.03 (d, J = 2 Hz, Ar, 2H), 7.10 (d, J = 2 Hz, Ar, 2H), 7.45 (d, J = 16.4 Hz, 2H); 13C NMR δ 14.88, 27.47, 60.81, 64.47, 111.93, 124.05, 126.35, 129.66, 132.43, 143.21, 149.27, 152.31, 198.29; Anal. Calcd. for C26H30O6: C, 71.21; H, 6.90; Found: C, 71.14; H, 6.96.
Figure 7
(3E,3’E)-4,4’-(5,5’-diethoxy-6,6’-dimethoxy-[1,1’-biphenyl]-3,3’-diyl)bis(but-3-en-2-one) (15)
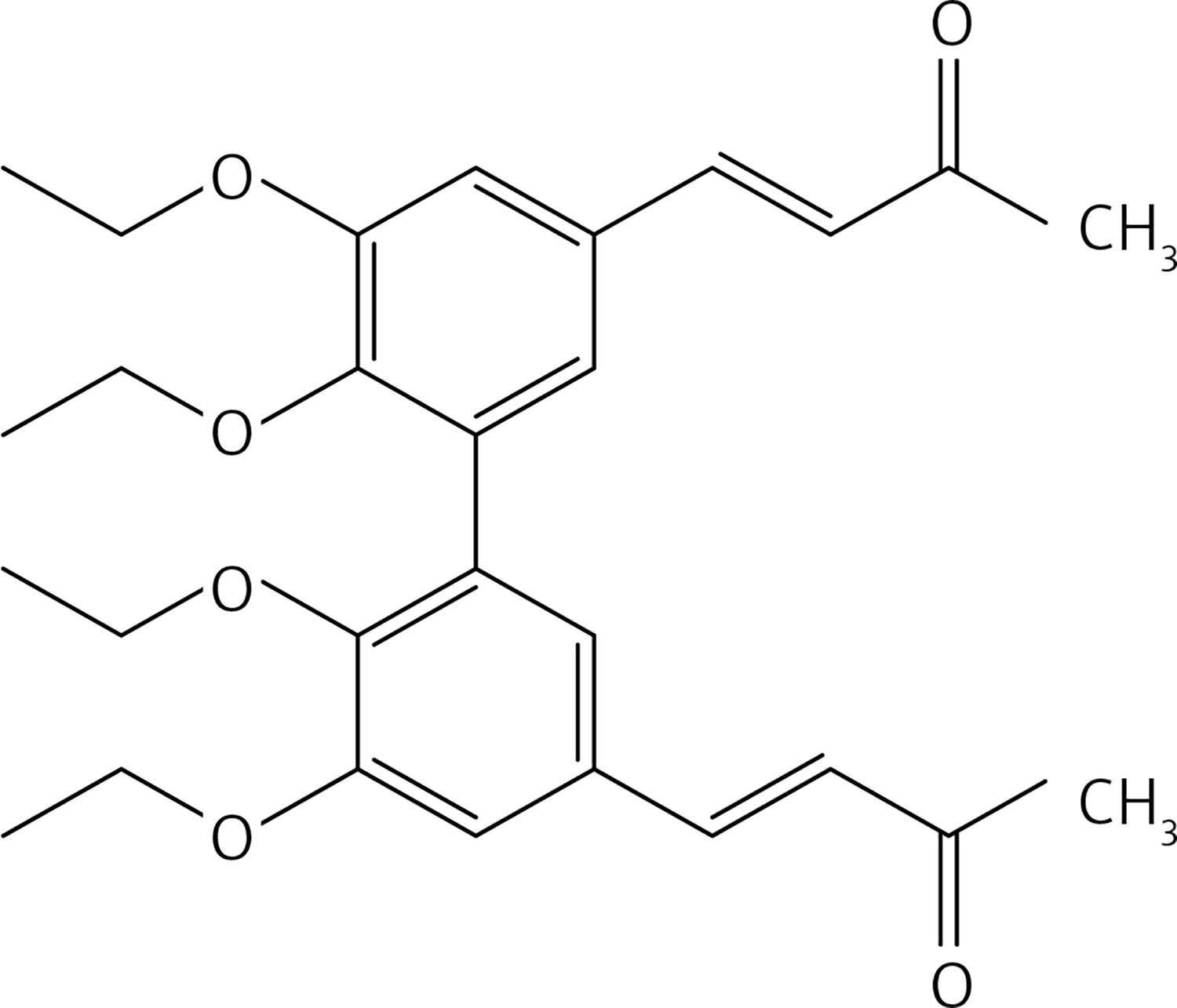
From 7, compound 17 (Figure 8) was obtained after flash chromatography (ethyl acetate : petroleum ether 1 : 2) as a yellow solid (0.82 g, 89%): mp 138–139°C; 1H NMR δ 1.09 (t, J = 7.2 Hz, 6H), 2.36 (s, 6H), 3.87-4.01 (series of m, 10H), 6.62 (d, J = 16 Hz, 2H), 7.10 (m, Ar, 4H), 7.45 (d, J = 16 Hz, 2H); 13C NMR δ 15.56, 27.50, 55.94, 69.16, 110.60, 124.41, 126.29, 129.50, 132.79, 143.21, 148.39, 153.22, 198.29; Anal. Calcd. for C26H30O6: C, 71.21; H, 6.90; Found: C, 71.29; H, 6.86.
Figure 8
(3E,3’E)-4,4’-(6,6’-diethoxy-5,5’-dimethoxy-[1,1’-biphenyl]-3,3’-diyl)bis(but-3-en-2-one) (17)
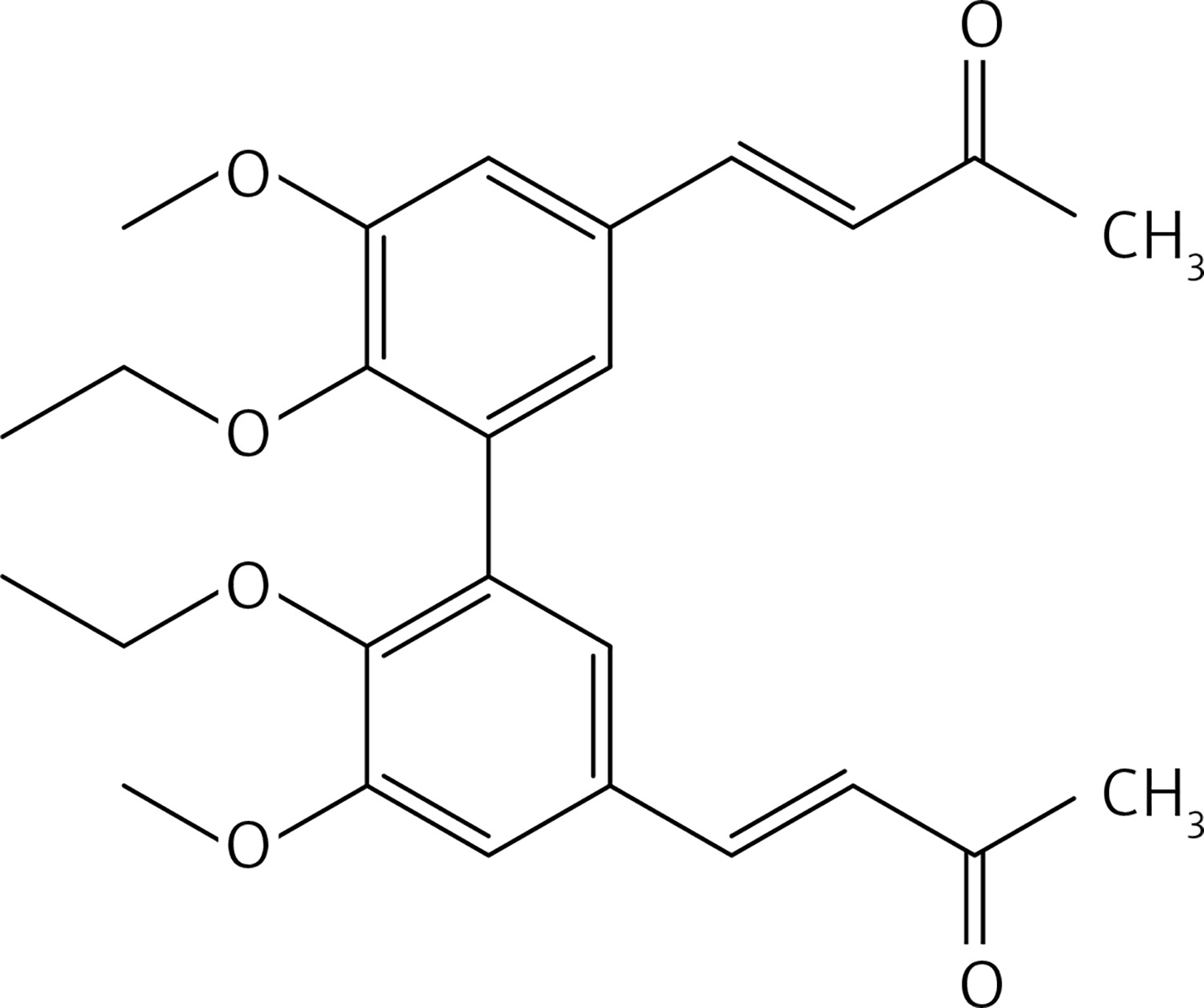
From 13, compound 19 (Figure 9) was obtained after flash chromatography (ethyl acetate : petroleum ether 2 : 3) as a yellow solid (0.78 g, 80%): mp 143–144°C; 1H NMR δ 1.11 (t, J = 7.2 Hz, 6H), 1.49 (t, J = 7.2 Hz, 6H), 2.35 (s, 6H), 3.92 (q, J = 7.2 Hz, 4H), 4.13 (q, J = 7.2 Hz, 4H), 6.63 (d, J = 16 Hz, 2H), 7.08 (d, J = 2.4 Hz, Ar, 2H), 7.10 (d, J = 2.4 Hz, Ar, 2H), 7.44 (d, J = 16 Hz, 2H); 13C NMR δ 14.88, 15.60, 27.47, 64.45, 69.14, 111.79, 124.38, 126.17, 129.34, 132.86, 143.35, 148.65, 152.48, 198.32; Anal. Calcd. for C28H34O6: C, 72.08; H, 7.35; Found: C, 72.14; H, 7.36.
From 25, compound 40 (Figure 10) was obtained after flash chromatography (acetone : petroleum ether 1 : 2) as a white solid (0.64 g, 56%): mp 162°C; 1H NMR δ 4.82 (m, 4H), 5.31 (dd, J = 1.6; 12.4 Hz, 2H), 5.51 (dd, J = 1.6, 18.8 Hz, 2H), 6.16 (m, 2H), 7.31(d, J = 9.2, Ar, 2H), 7.92 (dd, J = 2.4, 9.2 Hz, Ar, 2H), 7.98 (d, J = 2.4 Hz, Ar, 2H), 10.54 (s, 1H); 13C NMR δ 69.25, 114.30, 117.20, 125.23, 132.08, 133.00, 133.74, 160.36, 188.44, 205.27; Anal. Calcd. for C20H18O4: C, 74.52; H, 5.63; Found: C, 74.56; H, 5.66.
Methyl-tributylammonium permanganate (MTBAP) [31] (0.49 g, 1.5 mmol) in dry dichloromethane (15 ml) was added at room temperature, dropwise and under N2 to a solution of 22 (0.5 g, 3 mmol) in dry dichloromethane (20 ml). The reaction mixture was stirred at 20°C for 1 h and then was washed with an aqueous solution of Na2S2O5 (50 ml). The organic layer was separated, washed with water, dried over Na2SO4 and rotoevaporated to give a brown solid. Purification by flash chromatography using a 1 : 1 mixture of ethyl acetate : petroleum ether as eluent gave 23 (Figure 11) as a white solid (0.22 g, 46%): mp 83–84°C; 1H NMR δ 2.14 (s, 6H), 2.70-2.01 (series of m, 8H), 6.88 (d, J = 7.6 Hz, Ar, 2H), 7.10 (dd, J = 2.4, 7.6 Hz, Ar, 2H), 7.13 (d, J = 2.4 Hz, Ar, 2H); 13C NMR δ 28.08, 30.15, 45.35, 116.96, 125.24, 129.21, 131.22, 133.63, 151.21, 209.51; Anal. Calcd. for C20H22O4: C, 73.60; H, 6.79; Found: C, 73.66; H, 6.74.
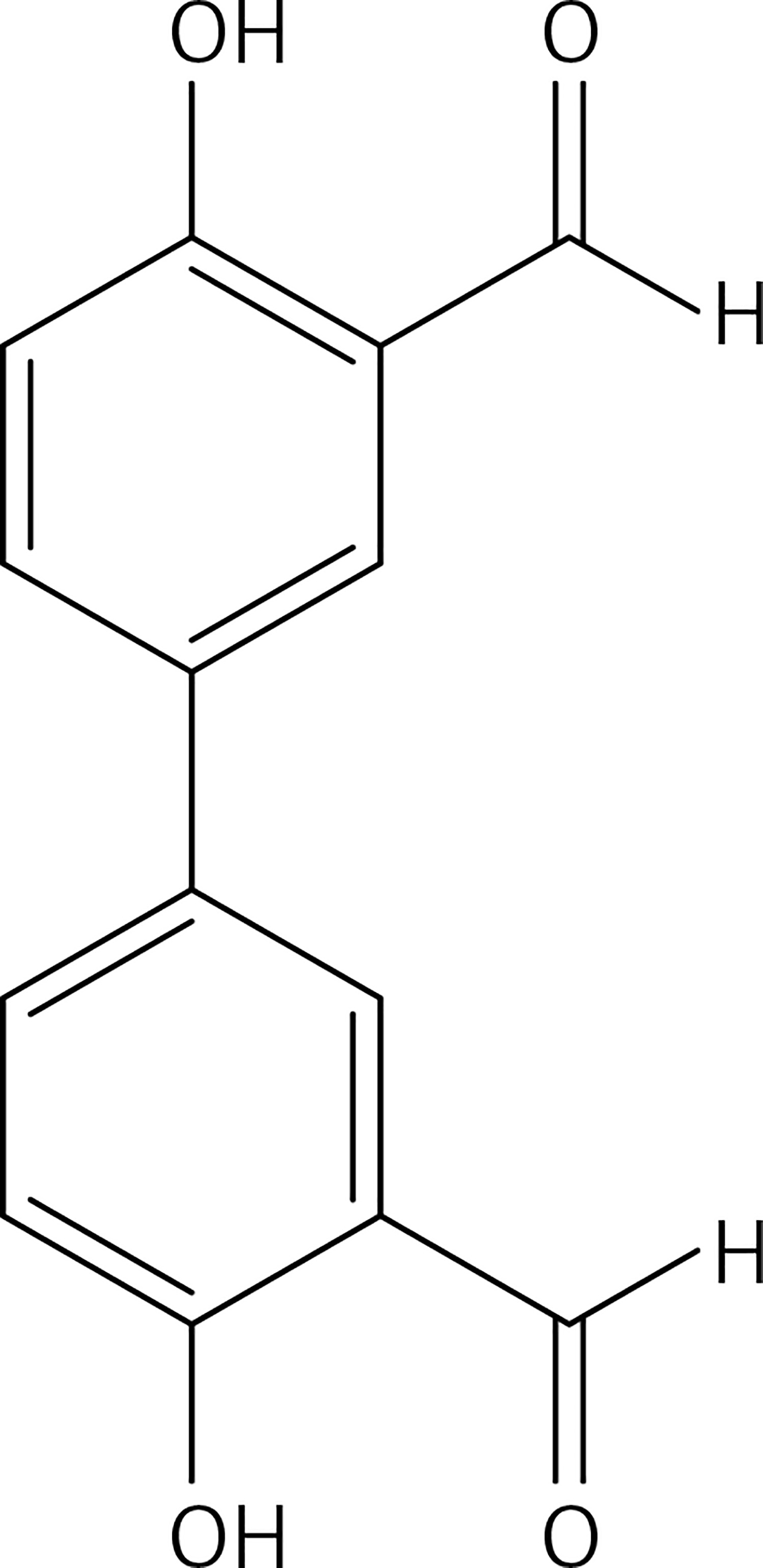
A solution of 4,4′-biphenol (0.20 g, 1.1 mmol) and hexamethylenetetramine (0.2 g, 1.3 mmol) in TFA (3 ml) was heated at 100°C for 10 min under microwave irradiation (100 W). The reaction mixture was extracted with dichloromethane (2 × 30 ml); the organic phase was washed with NaHCO3 solution, dried over Na2SO4 and evaporated. The crude product was washed with EtOH to eliminate impurities to give compound 25 (Figure 12) as a yellow solid (0.16 g, 60%): mp 106–107°C (Lit.53 106–108°C); 1H NMR (acetone d6) δ 7.10 (d, J = 8.8 Hz, 2H), 7.93 (dd, J = 2.4 and 8.8 Hz, 2H), 8.11 (d, J = 2.4 Hz, 2H), 10.13 (s, 2H); 13C NMR (acetone d6): δ 117.83, 121.24, 131.22, 131.30, 134.80, 160.59, 197.09; Anal. Calcd. for C14H10O4: C, 69.42; H, 4.16; Found: C, 69.45; H, 4.19.
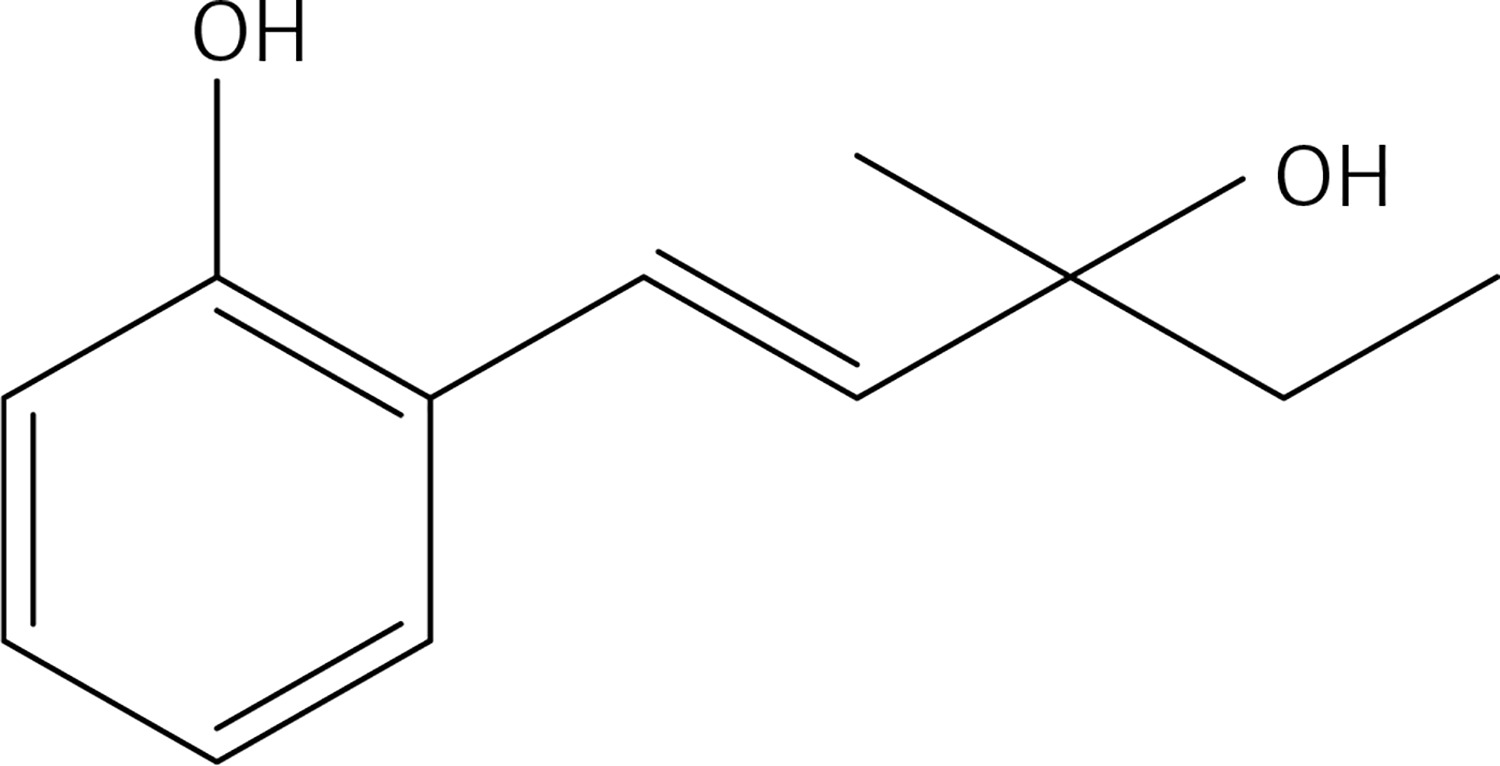
Cs2CO3 (0.47 g, 1.5 mmol), Pd (OAc)2 (0.06 g, 0.29 mmol) and 3-methylpent-1-en-3-ol (0.58 g, 5.8 mmol) were added to a solution of 2-bromophenol (0.5 g, 2.9 mmol) in DMF (10 ml). The mixture was stirred under MW irradiation for 20 min at 100 W and 100°C. The reaction mixture was diluted with Et2O and washed with brine. The organic layer was dried over Na2SO4, and rotoevaporated. The product was purified by flash chromatography on silica gel using a 1 : 3 mixture of ethyl acetate: petroleum ether as eluent to give product 26 (Figure 13) (0.23 g, 42%): mp 108–109°C; 1H NMR (acetone d6) δ 0.91 (t, J = 7.6 Hz, 3H), 1.31 (s, 3H); 1.62 (q, J = 7.6 Hz, 2H); 2.84 (bs, OH); 6.31 (d, J = 16.4 Hz, 1H); 6.8 (td, J = 0.8, 7.2 Hz, 1H); 6.86 (dd, J = 0.8, 8 Hz, 1H); 6.91 (d, J = 16.4 Hz, 1H); 7.03 (td, J = 1.2 and 8.0 Hz, 1H); 7.41 (dd, J = 1.2, 8 Hz, 1H), 8.37 (bs, OH); 13C NMR δ 8.36, 27.69, 35.38, 51.70, 115.92, 120.82, 121.77, 124.22, 127.30, 28.51, 129.01, 138.04; Anal. Calcd. for C12H16O2: C, 74.97; H, 8.39; Found: C, 74.88; H, 8.43.
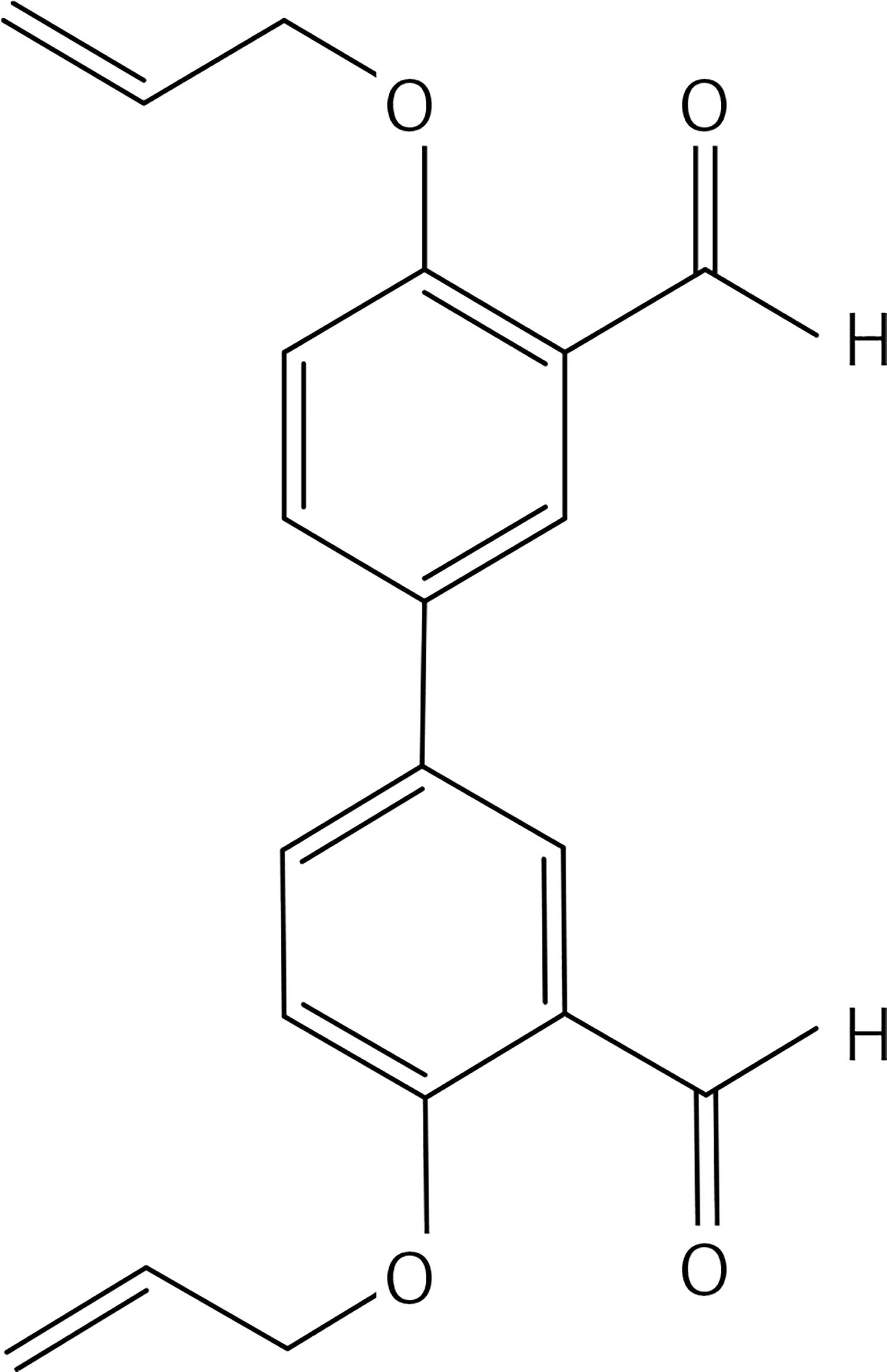
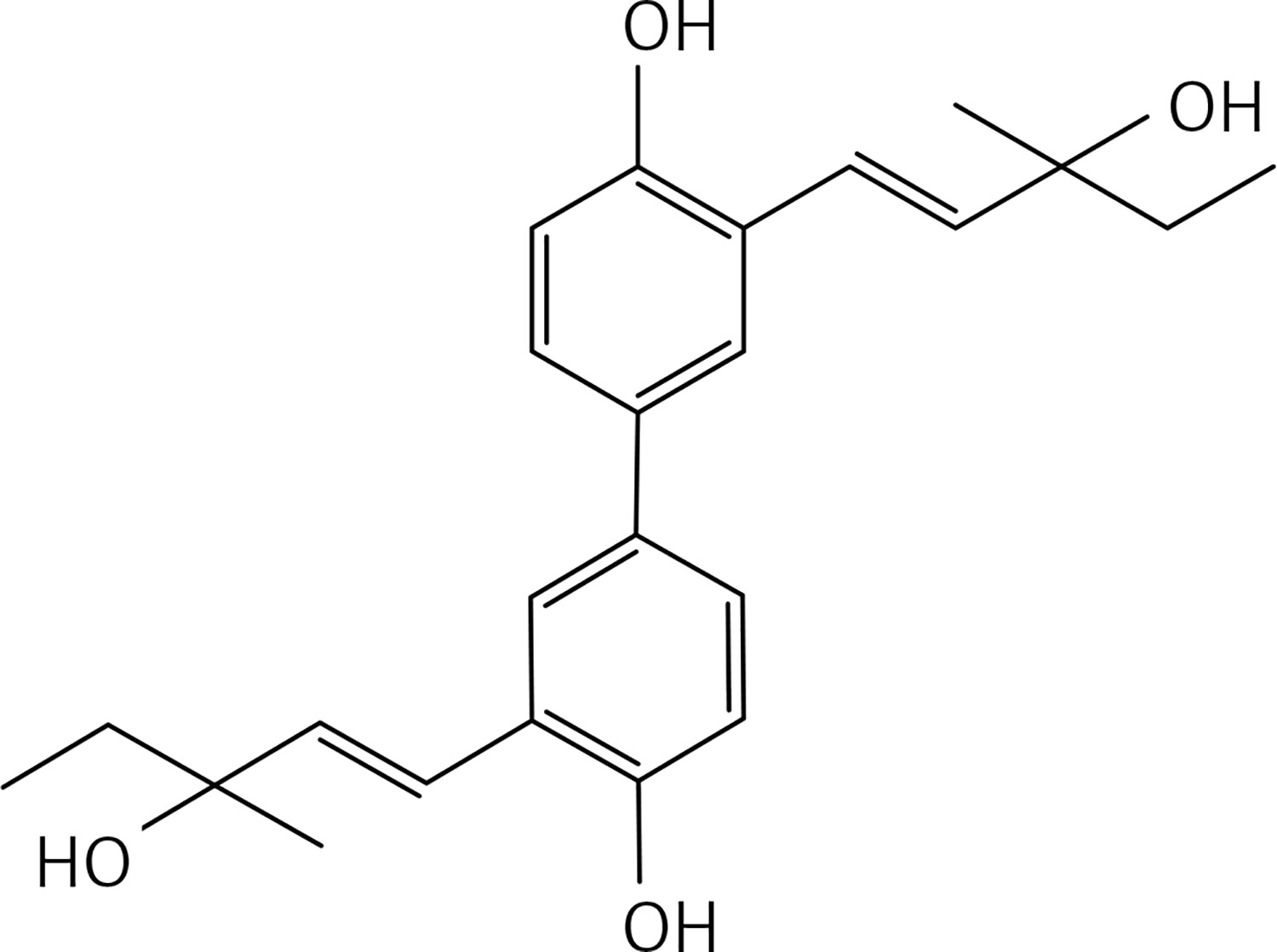
A solution of compound 42 (0.5 g, 1.45 mmol) in DMF (30 ml) Cs2CO3 (0.94 g, 2.9 mmol), Pd(OAc)2 (0.097 g, 0.145 mmol) and 3-methylpent-1-en-3-ol (0.58 g, 5.8 mmol) was stirred under MW irradiation at 100 W for 20 min and at 100°C. The reaction mixture was then diluted with Et2O and washed with brine. The organic layer, dried over Na2SO4, was concentrated under vacuum. The product was purified by flash chromatography on silica gel eluting with a 3 : 1 mixture of petroleum:ethylacetate to give product 27 (Figure 14) (0.2 g, 40%): mp 130°C; 1H NMR(acetone d6) δ 0.92 (t, J = 7.2 Hz, 4H), 1.32 (s, 6H), 1.62 (q, J = 7.2 Hz, 6H), 3.52 (bs, OH, 2H), 6.47 (d, J = 16.0 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 7.28 (dd, J = 2.0, 8.8 Hz, 2H), 7.64 (d, J = 2.0 Hz), 8.49 (bs, OH, 2H); 13C NMR δ 7.89, 27.37, 35.42, 72.35, 115.94, 121.61, 124.65, 124.73, 126.01, 132.79, 137.60, 153.55; Anal. Calcd. for C24H30O4: C, 75.36; H, 7.91; Found: C, 75.38; H, 7.96.
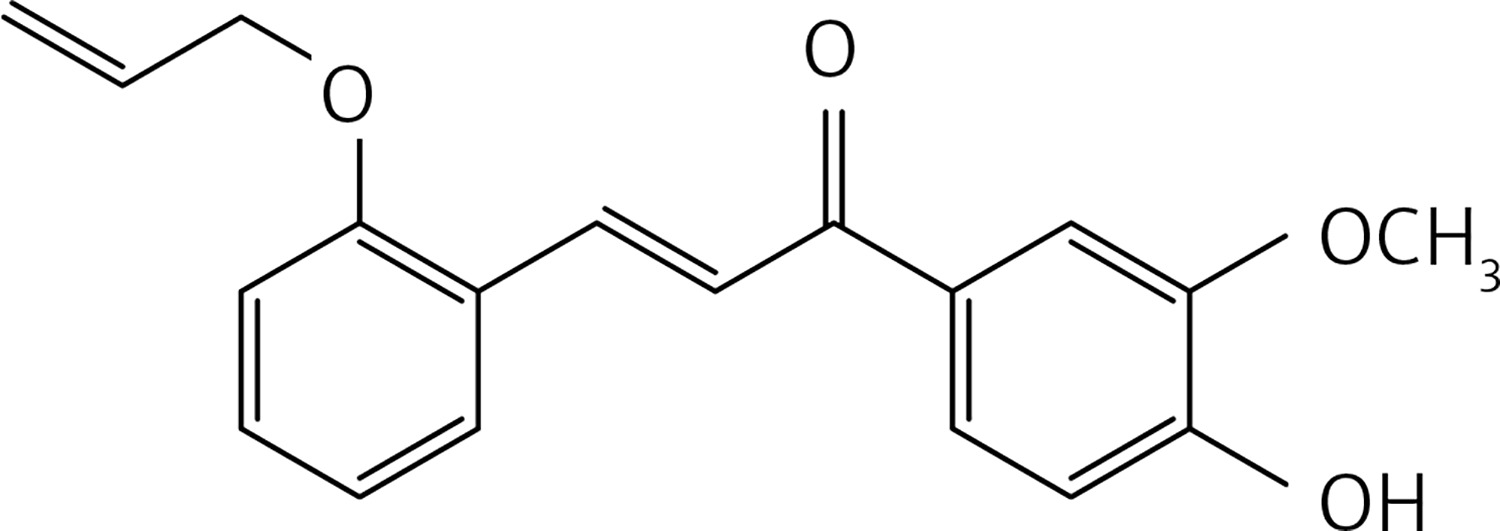
A mixture of 2-(allyloxy)benzaldehyde [32] (0.20 g, 1.2 mmol), apocynin (0.2 g, 1.2 mmol) and LiOH (0.17 g, 7.4 mmol) in MeOH (5 ml) was subjected, in a 30 ml glass pressure microwave tube, equipped with a magnetic stirrer bar, to microwave irradiation (power: 100 W; temperature: 70°C) for 15 min. The reaction mixture was then acidified with HCl 10% solution and extracted with dichloromethane (2 × 30 ml). The organic solution was dried over Na2SO4 and roto-evaporated. The product was purified by flash chromatography using a 4 : 1 petroleum ether : acetone mixture as eluent, to obtain compound 28 (Figure 15) as a yellow solid (0.2 g, 53%): mp 94°C; 1H NMR δ 3.96 (s, 3 H), 4.63 (d, J = 5.2 Hz, 2H), 5.44 (dd, J = 1.2, 17.2 Hz, 2H), 6.10 (m, 1H), 6.91 (d, J = 8.0 Hz, Ar, 1H), 6.97 (m, Ar, 2H), 7.34 (t, J = 1.6 Hz, Ar, 1H), 7.65 (m, Ar, 3H), 7.71 (d, J = 16 Hz, 1H), 8.14 (d, J = 16 Hz, 1H). 13C NMR δ 56.10, 69.10, 110.53, 112.49, 113.72, 117.99, 120.94, 122.72, 123.64, 124.34, 129.57, 131.25, 131.41, 132.87, 139.67, 146.49, 150.19, 157.75, 189.22; Anal. Calcd. for C19H18O4: C, 73.53; H, 5.85; Found: C, 73.56; H, 5.86.
A mixture of compound 40 (0.26 g, 0.82 mmol), apocynin (0.29 g, 1.8 mmol) and LiOH (0.41 g, 9.8 mmol) in MeOH (10 ml) was subjected to microwave irradiation (power: 70 W; temperature: 70°C) for 30 min in a 30 ml glass pressure microwave tube. The reaction mixture was then acidified with HCl 10% solution and extracted with dichloromethane (2 × 30 ml). The organic phase was dried over Na2SO4 and evaporated. Crude purification by flash chromatography using a 2 : 1 petroleum ether : acetone mixture as eluent gave product 29 (Figure 16) as a yellow solid (0.2 g, 40%): mp 204°C; 1H NMR δ 3.93 (s, 6H), 4.78 (m, 4H), 5.33–5.37 (series of m, 2H), 5.51–5.56 (series of m, 2H), 6.22 (m, 2H), 6.95 (d, J = 8.4 Hz, Ar, 2H), 7.20 (d, J = 8.8 Hz, Ar, 2H), 7.70 (d, J = 1.2 Hz, Ar, 2H), 7.73 (dd, J = 1.2; 10.8 Hz, Ar, 2H), 7.81 (dd, J = 1.2; 10.0 Hz, Ar, 2H), 8.05 (d, J = 16 Hz, 2H), (d, J = 8.8 Hz, Ar, 2H), 8.2 (d, J = 16 Hz, 2H), 8.22(d, J = 2.4 Hz, Ar, 2H); 13C NMR δ 55.43, 69.13, 111.19, 113.18, 114.56, 117.12, 122.70, 123.46, 124.51, 126.88, 129.64, 130.80, 132.91, 133.52, 137.84, 147.66, 151.42, 156.94, 187.37; Anal. Calcd. for C38H34O8: C, 73.77; H, 5.54; Found C, 73.80, H, 5.57.
Figure 16
(E)-3-(4,4’-bis(allyloxy)-3’-((E)-3-(3-hydroxy-4-methoxyphenyl)-3-oxoprop-1-en-1-yl)-[1,1’-biphenyl]-3-yl)-1-(4-hydroxy-3-methoxyphenyl) prop-2-en-1-one (29)
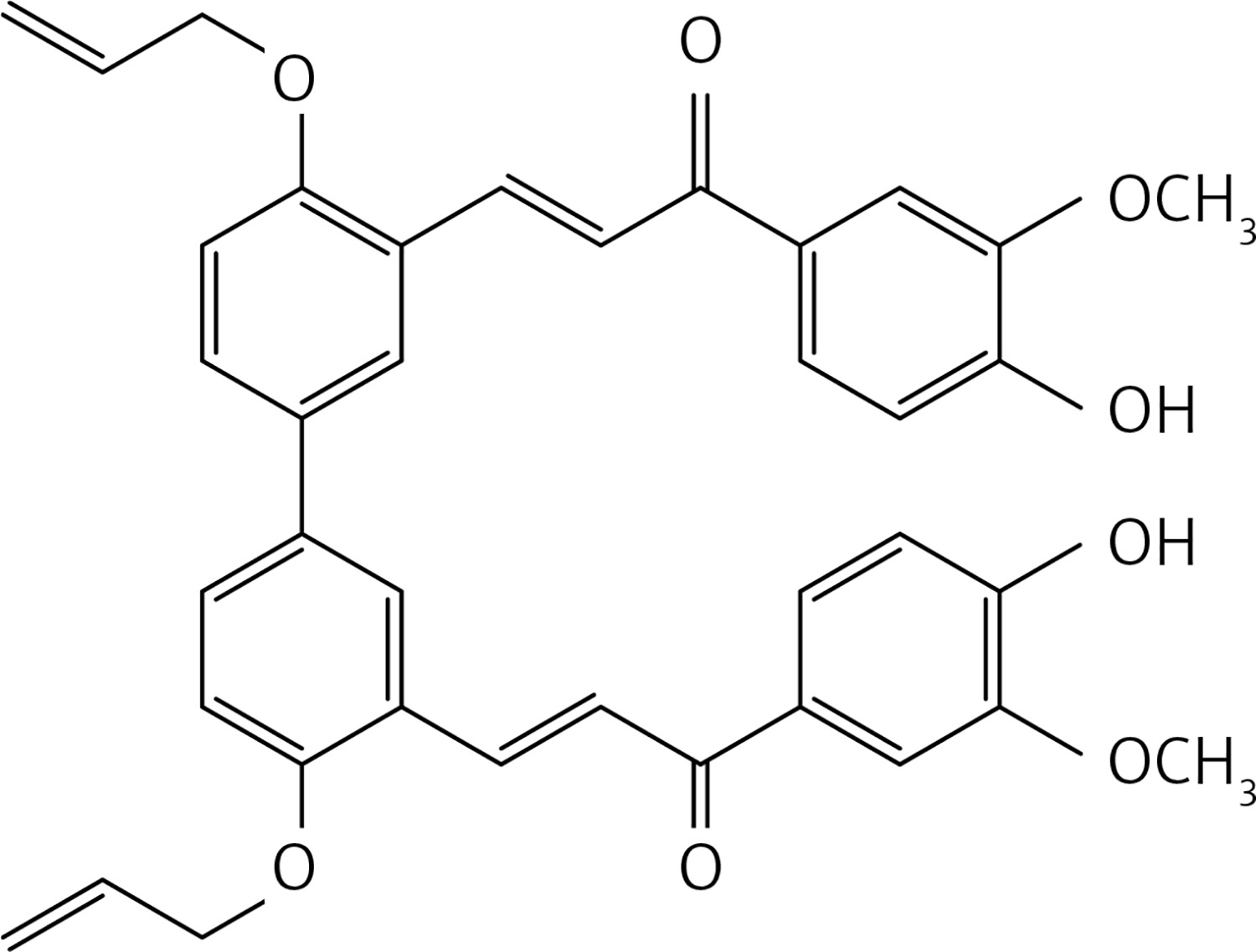
To a solution of 2 (allyloxy) benzaldehyde [32] (1 g, 6.2 mmol) and 3,4-dimethoxy acetophenone (1.1 g, 6.2 mmol) in MeOH (10 ml) a solution of LiOH (0.89 g, 37.2 mmol) in MeOH (5 ml) was added dropwise. The reaction mixture was stirred at reflux for 12 h. The solution was acidified with HCl 10% solution and extracted with dichloromethane (2 × 30 ml). The organic phases were dried over Na2SO4 and concentrated in vacuo. The crude product was purified by flash chromatography using dichloromethane as eluent to give product 30 (Figure 17) as a white solid (1.25 g, 62%): mp 87–88°C; 1H NMR δ 3.95 (s, 3 H), 3.96 (s, 3 H), 4.61 (d, J = 5.2 Hz, 2H), 5.26-5.47 (series of m, 2H), 6.08 (m, 1H), 6.88 (d, J = 8.0 Hz, Ar, 1H), 6.97 (t, J = 6.8 Hz, Ar, 1H), 7.31 (dt, J = 1.6, 4.8 Hz, Ar, 1H), 7.60-7.68 (series of m, Ar, 4H), 7.70 (d, J = 16 Hz, 1H), 8.10 (d, J = 16 Hz, 1H). 13C NMR δ 55.97, 56.05, 69.14, 109.97, 110.79, 112.46, 117.96, 120.92, 122.64, 122.97, 124.29, 129.58, 131.42, 131.54, 132.86, 139.65, 149.07, 153.02, 157.03, 189.14; Anal. Calcd. for C20H20O4: C, 74.06; H, 6.22; Found: C, 74.16; H, 6.29.
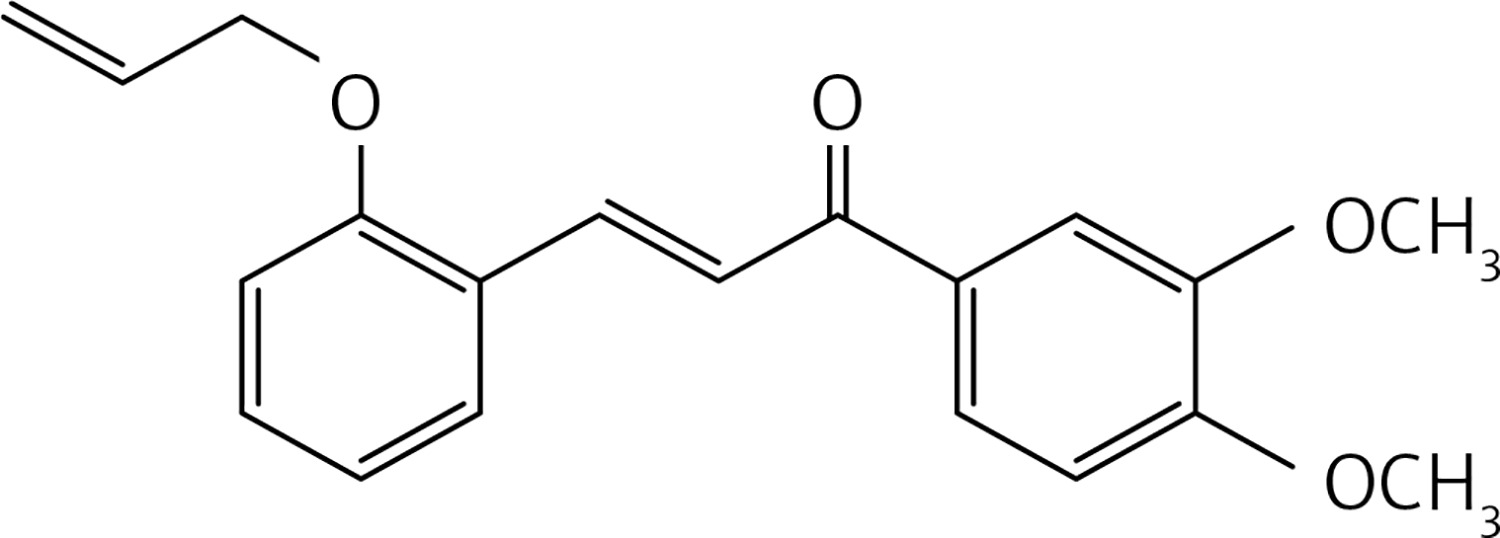
In a glass pressure microwave tube, a mixture of compound 40 (0.5 g, 1.6 mmol), 3,4-dimethoxyacetophenone (0.6 g, 3.4 mmol) and LiOH (0.78 g, 18.6 mmol) in MeOH (10 ml) was subjected to microwave irradiation (power: 70 W; temperature: 70°C) and stirred for 30 min. The reaction mixture was then acidified with HCl 10% solution and extracted with dichloromethane (2 × 30 ml). The organic solution was dried over Na2SO4 and evaporated. Purification by flash chromatography using a 7 : 3 petroleum ether : acetone mixture as eluent gave product 31 (Figure 18) as a yellow solid (0.16 g, 31%): mp 145–146°C; 1H NMR δ 3.95 (s, 3H), 3.97 (s, 3H), 4.70 (d, J = 5.6 Hz, 4H), 5.33–5.57 (series of m, 4H), 6.22 (m, 2H), 6.94 (d, J = 8.0 Hz, Ar, 2H), 7.02 (d, J = 8.0 Hz, Ar, 2H), 7.54 (dd, J = 2, 8.4 Hz, Ar, 2H), 7.64 (m, Ar, 2H), 7.71 (dd, J = 1.2, 8.4 Hz, Ar, 2H), 7.79 (d, J = 16 Hz, 2H), 7.82 (d, J = 2 Hz, Ar, 2H), 8.14 (d, J = 16 Hz, 2H), 8.22(d, J = 2.4 Hz, Ar, 2H); 13C NMR δ 56.04, 56.07, 69.42, 110.01, 110.85, 112.95, 118.16, 123.09, 123.20, 124.71, 127.97, 129.65, 131.51, 132.80, 133.16, 139.58, 149.17, 153.14, 157.13, 189.16; Anal. Calcd. for C40H38O8: C, 74.29; H, 5.92; Found C, 74.35, H, 5.87.
Figure 18
(2E,2’E)-3,3’-(4,4’-bis(allyloxy)-[1,1’-biphenyl]-3,3’-diyl)bis(1-(3,4-dimethoxyphenyl)prop-2-en-1-one) (31)
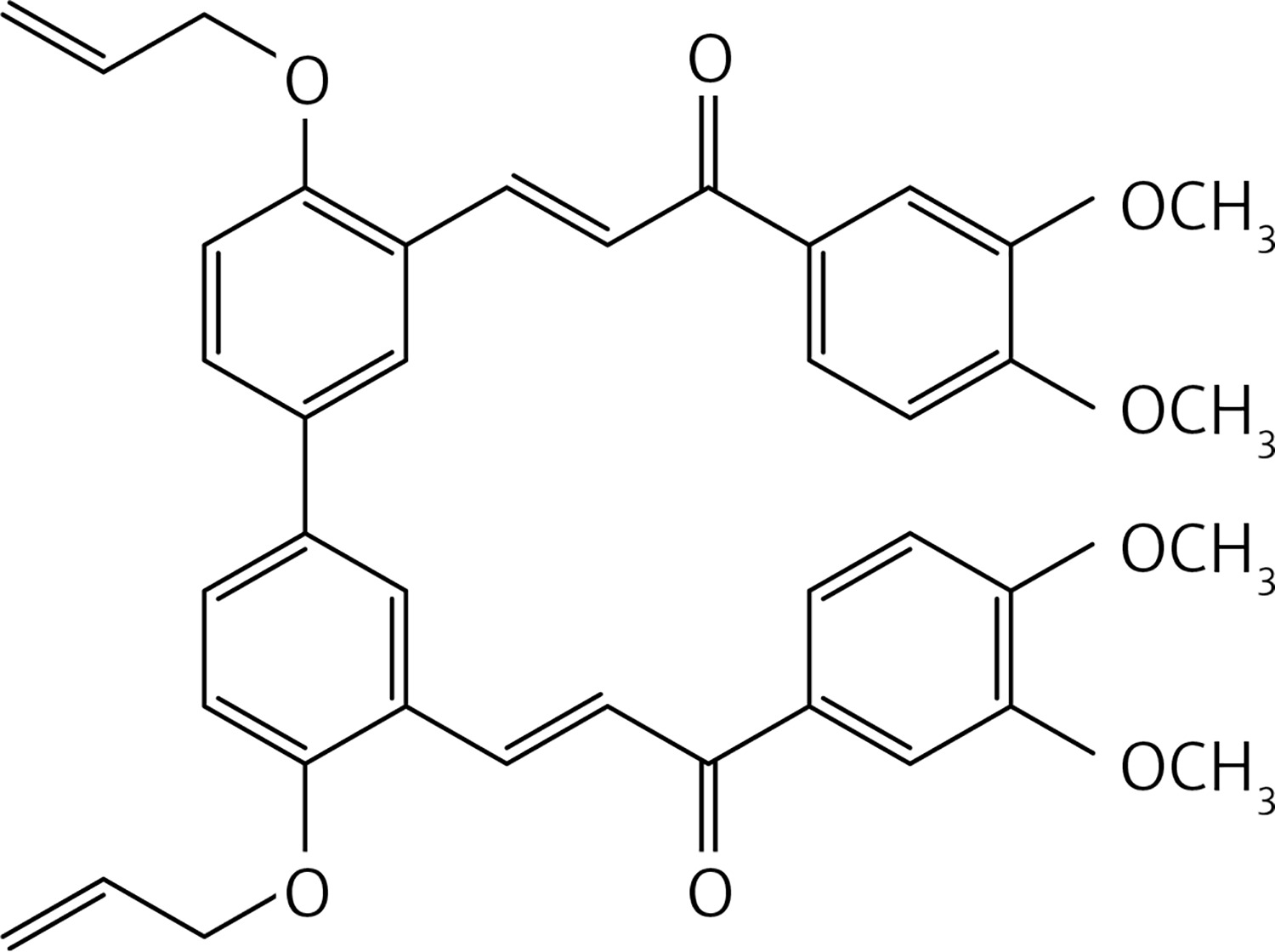
In a glass pressure microwave tube, ethyl vanillin (0.1 g, 0.6 mmol), iron sulfate heptahydrate (0.0075 g, 0.03 mmol) and potassium peroxodisulfate (0.081 g, 0.3 mmol) in water (5 ml) were subjected, with stirring, to microwave irradiation (power: 100 W; temperature: 110°C) for 10 min. The reaction mixture was then acidified with HCl 10% solution and extracted with dichloromethane (2 × 30 ml). The organic solution was dried over Na2SO4 and evaporated. The product was purified by chromatography on a column of silica gel with a mixture of 1 : 4 petroleum ether : ethyl acetate as eluent to give compound 39 (Figure 19) as a yellow solid (0.51 g, 52%): mp 236–237°C; 1H NMR δ 1.49 (t, J = 6.8 Hz, 6H), 4.23 (q, J = 6.8 Hz, 4H), 6.6 (bs, 2H), 7.42 (d, J = 1.6 Hz, Ar, 2H), 7.50 (d, J = 1.6 Hz, Ar, 2H); 13C NMR δ 14.71, 65.13, 108.71, 113.89, 120.76, 129.26, 131.48, 143.23, 190.48; Anal. Calcd. for C18H18O6: C, 65.45; H, 5.49; Found: C, 65.50; H, 5.59.
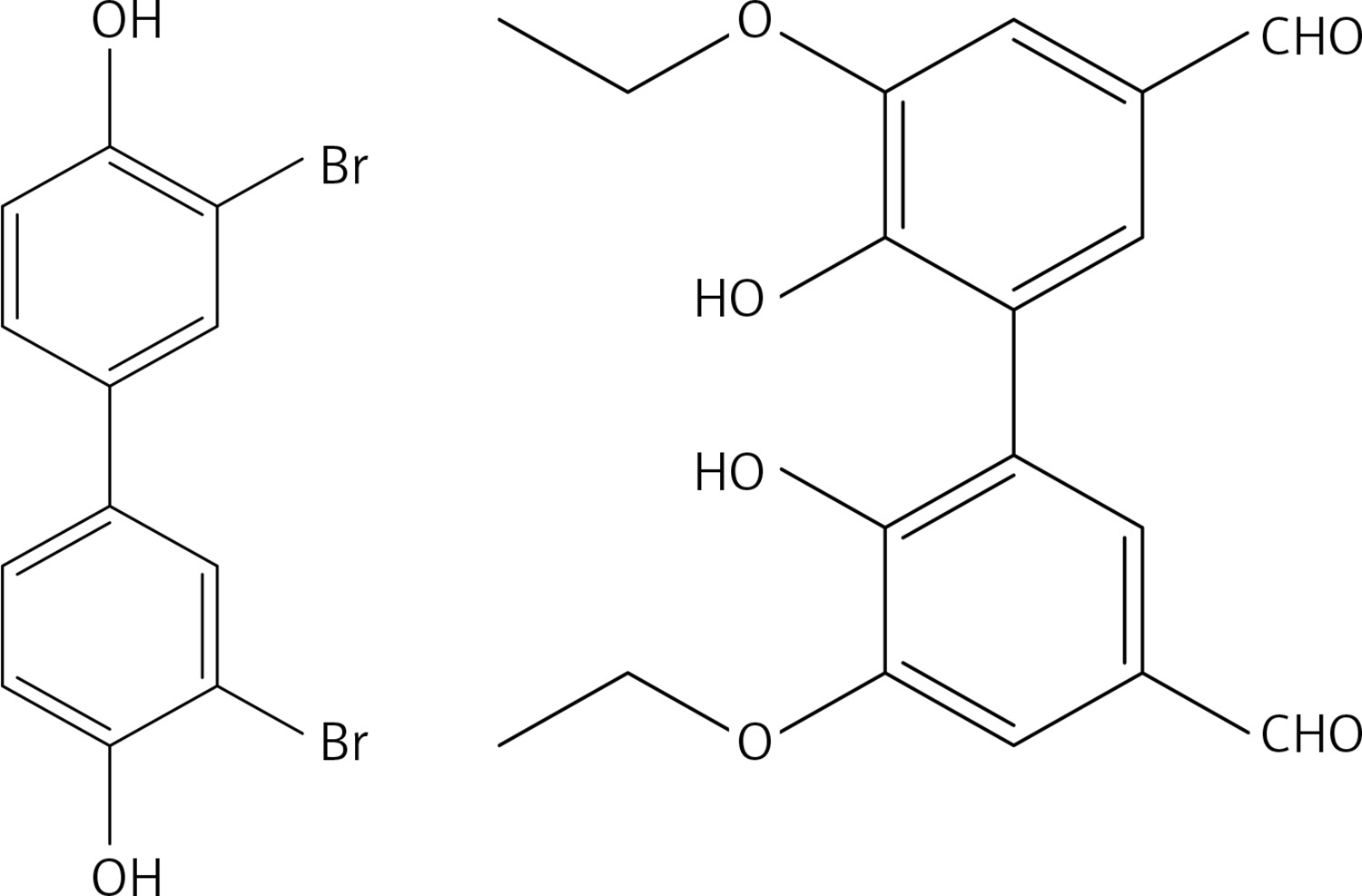
To a solution of 5,5’,6,6’-tetramethoxy-[1,1’-biphenyl]-3,3’-diol [14] (1.17 g, 3.8 mmol) and K2CO3 (2 g, 11.4 mmol) in dry DMF (30 ml), methyl bromoacetate (1.50 g, 8.4 mmol) was added. The mixture was stirred at rt for 72 h and then water and 10% HCl were added. The precipitate was filtered to obtain 43 (Figure 20) as a yellow solid (1.62 g, 95%): mp 132–133°C; 1H NMR δ 3.56 (s, 6H), 3.78 (s, 6H), 3.86 (s, 6H), 4.57 (s, 4H), 6.31 (d, J = 2.4 Hz, Ar, 2H), 6.61 (d, J = 2.4 Hz, Ar, 2H); 13C NMR δ 52.21, 55.84, 60.74, 65.60, 100.92, 106.34, 132.27, 141.53, 153.51, 153.54, 169.31; Anal. Calcd. for C22H26O10: C, 58.66; H, 5.82; Found: C, 58.67; H, 5.79.
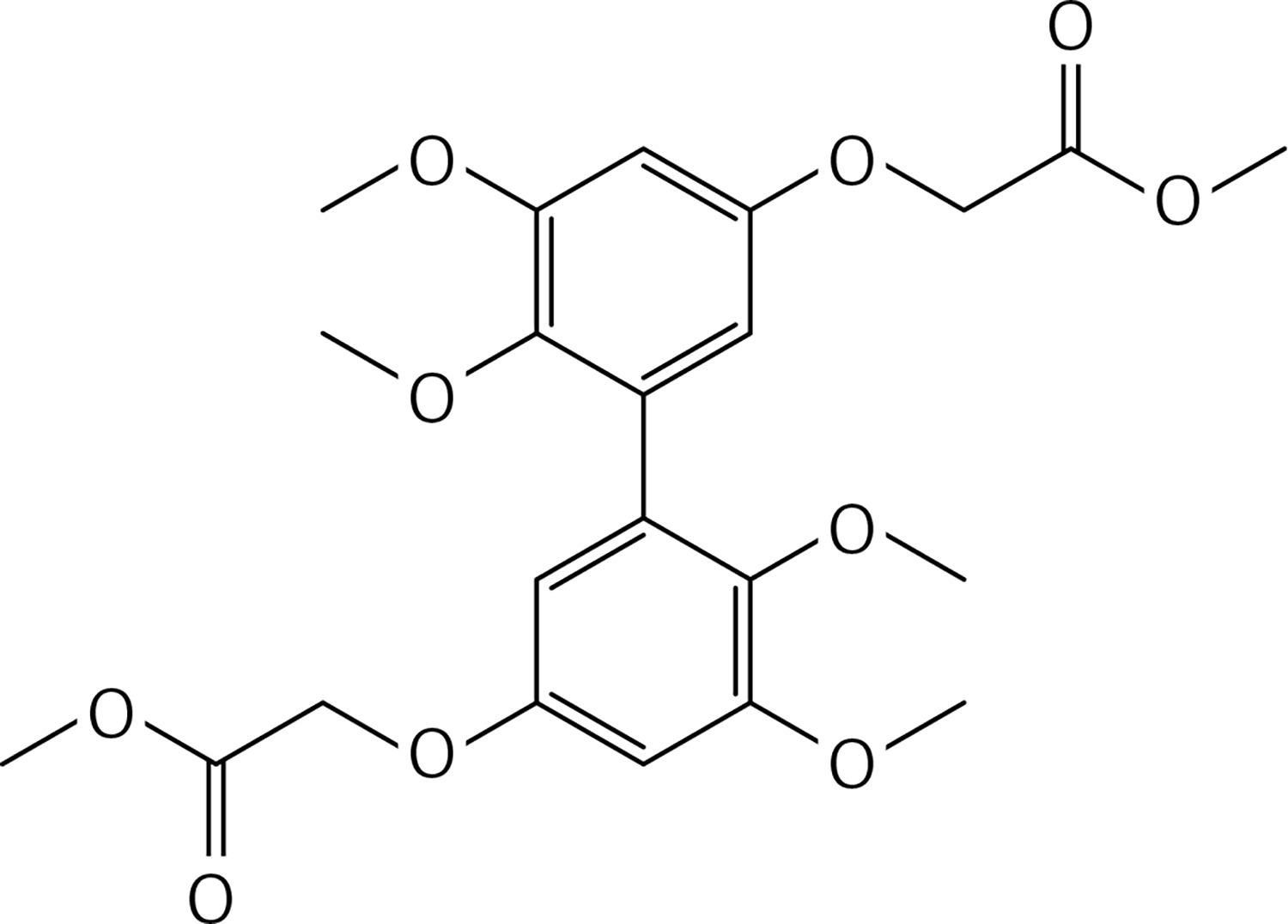
To a solution of 43 (1.32 g, 2.9 mmol in dry THF (25 ml), lithium hydroxide (0.50 g, 11.7 mmol) was added under N2. The solution was stirred for 12 h at 70°C. 10% HCl and water were added, the mixture was extracted with diethyl ether (2 ×50 ml). The organic phases were dried and evaporated to get 44 (Figure 21) as a white solid (1.20 g, 98%): mp 183–184°C; 1H NMR δ 3.52 (s, 6H), 3.83 (s, 6H), 4.67 (s, 4H), 6.37 (d, J = 2.1 Hz, Ar, 2H), 6.63 (d, J = 2.1 Hz, Ar, 2H); 13C NMR δ 55.26, 59.73, 64.97, 100.32, 106.99, 132.99, 141.36, 153.55, 153.81, 169.46; Anal. Calcd. for C20H22O10: C, 56.87; H, 5.25; Found: C, 56.79; H, 5.29.
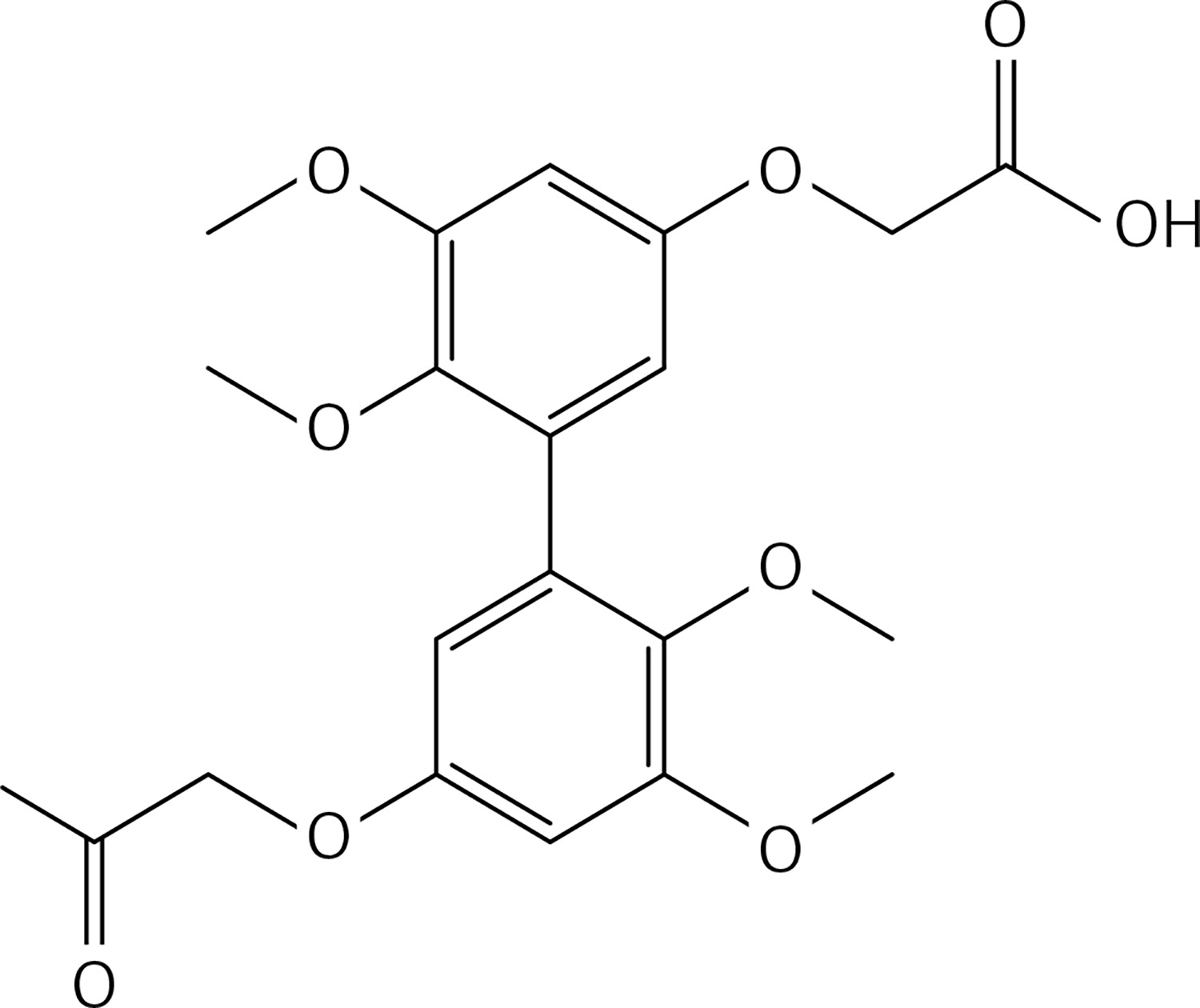
8 g of polyphosphoric acid was weighed in a 25 ml flask and heated at an oil bath temperature of 90°C. Within 5 min compound 44 (0.3 g, 0.7 mmol) was added; then stirring was carried out for 3 h at 90°C. The cooled solution was poured onto ice. After stirring for 2 h, three extractions were carried out with a total of 400 ml of dichloromethane. The organic phases, washed with water and 10% K2CO3 solution, were dried over Na2SO4. After removal of the solvent, the residue was purified by chromatography on a column of silica gel using a 1 : 1 mixture of ethyl acetate : petroleum ether as eluent to get 37 (Figure 22) as a yellow solid (0.24 g, 90%): mp 138–140°C; 1H NMR δ 3.59 (s, 6H), 3.92 (s, 6H), 4.52 (AB system, J = 17.6 Hz, 4H), 6.65 (s, Ar, 2H); 13C NMR δ 56.24, 60.96, 75.45, 96.07, 111.35, 124.01, 142.56, 161.24, 172.43, 196.85; Anal. Calcd. for C20H18O8: C, 62.18; H, 4.70; Found: C, 62.14; H, 4.56.
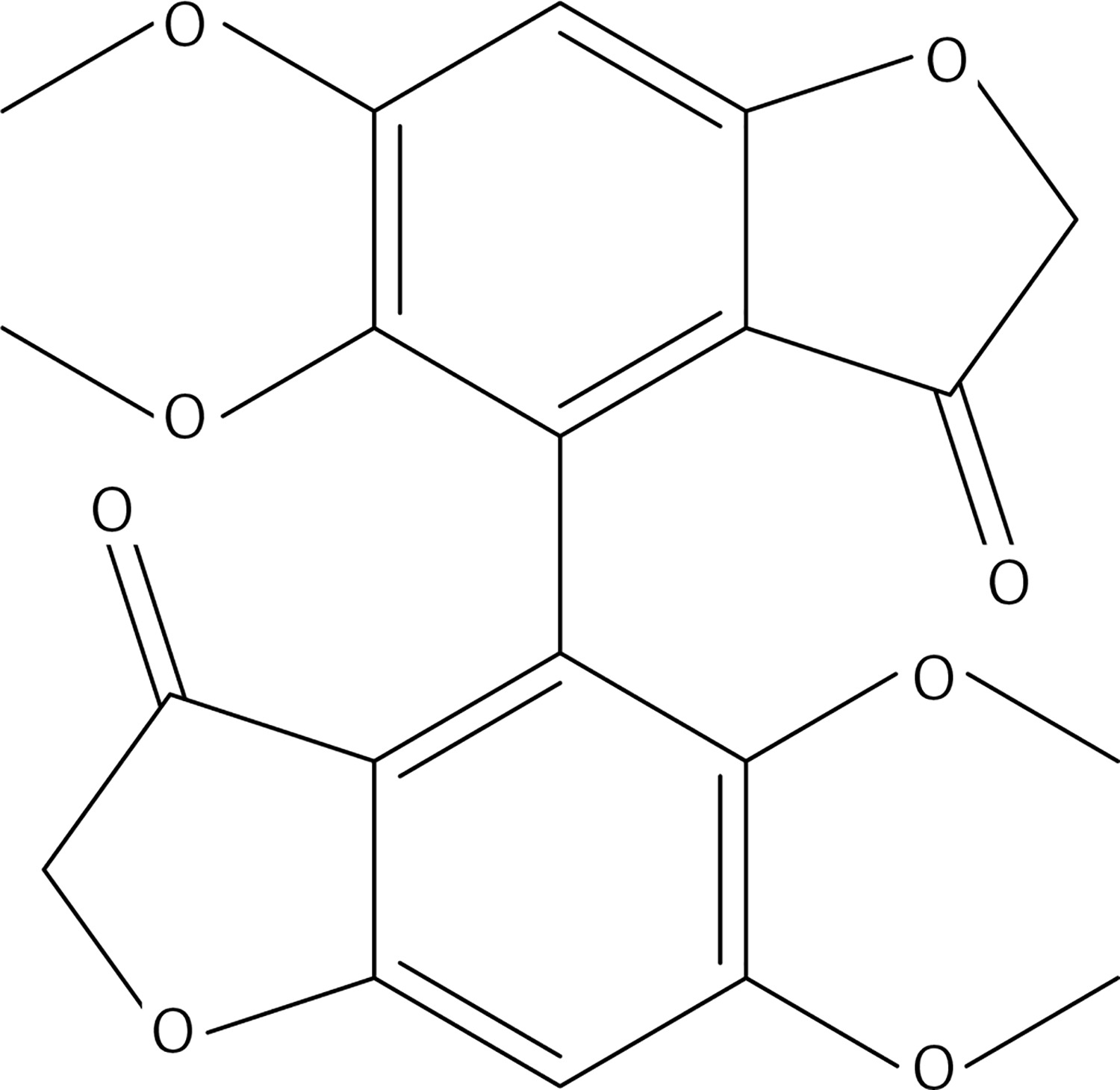
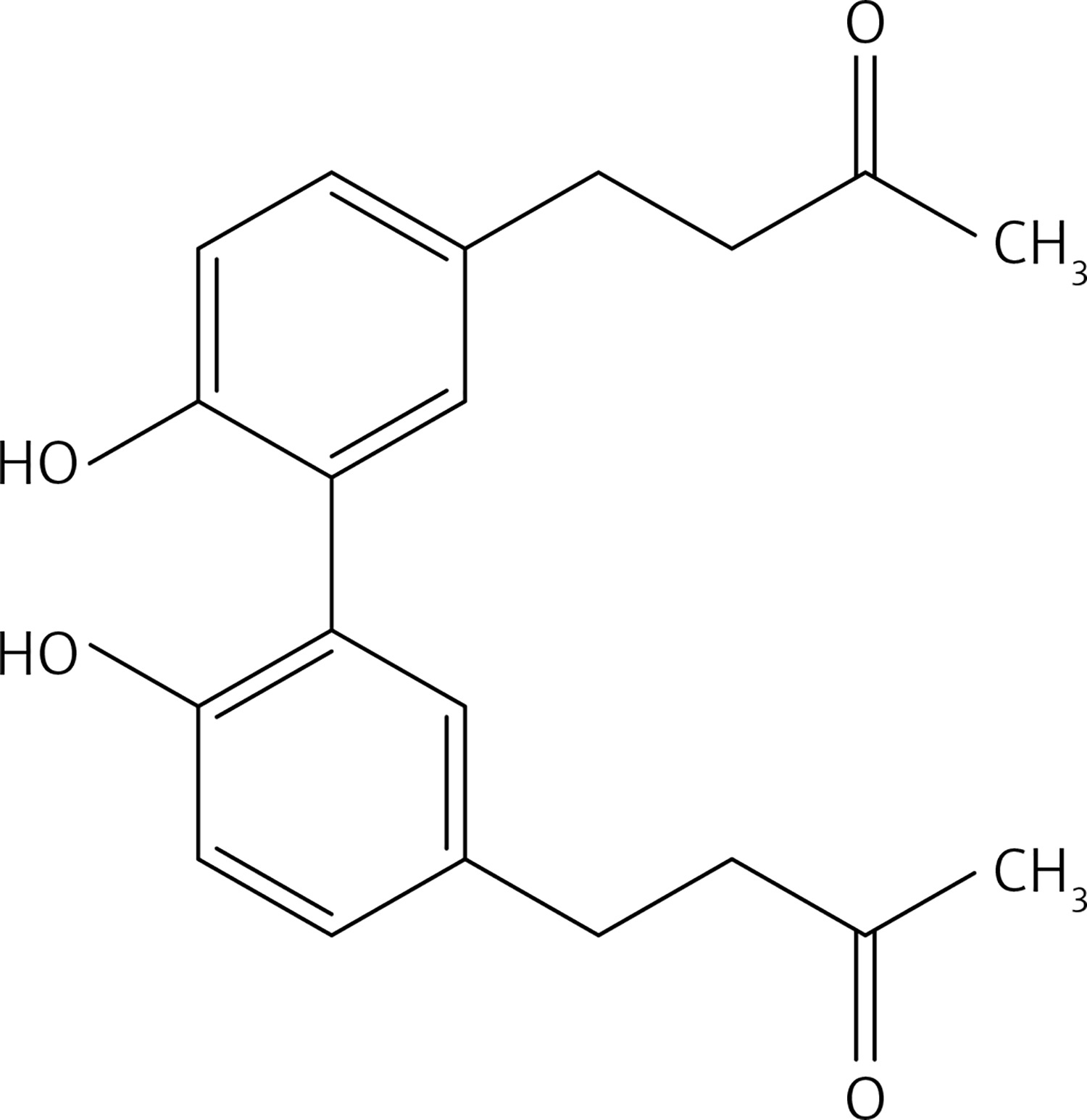
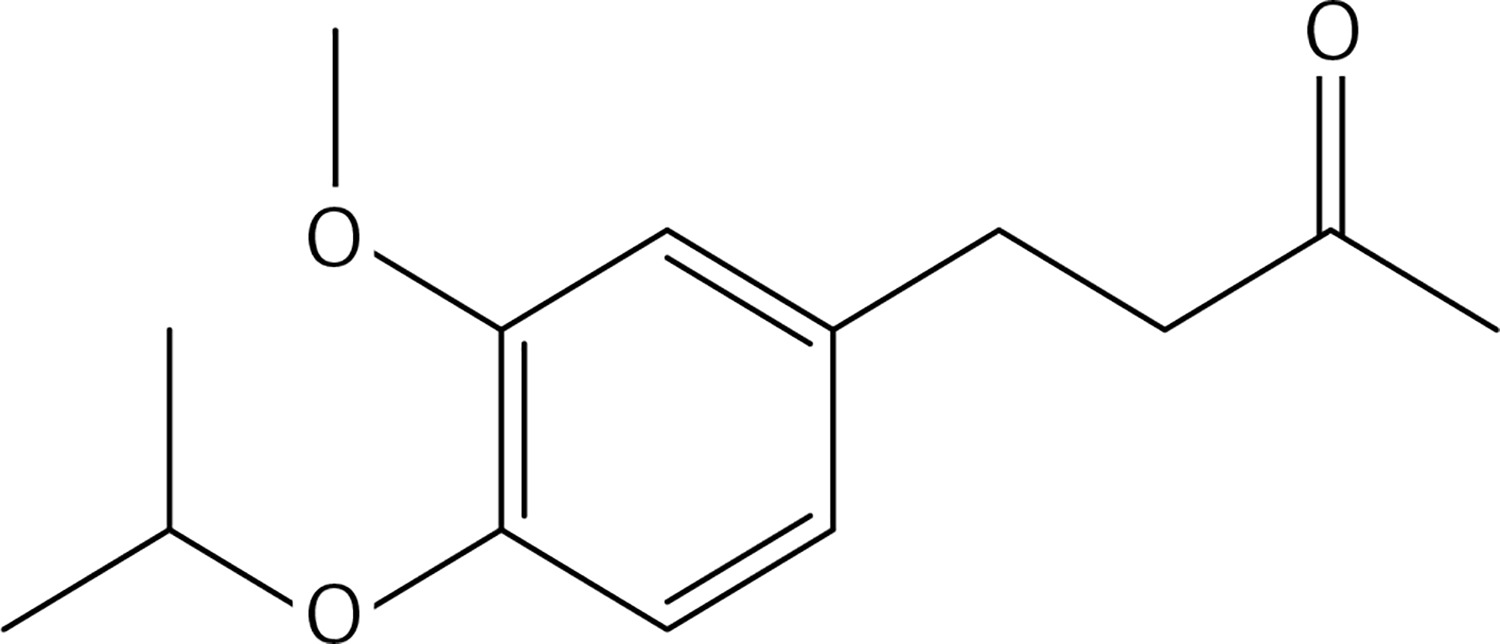
2-bromopropane (1.5 g, 12.4 mmol) was added to a mixture of compound 3 (2 g, 10.3 mmol) and K2CO3 (1.7 g, 12.4 mmol) in dry acetone (30 ml). The reaction mixture was stirred at 60°C for 12 h, filtered and evaporated. The resulting solid was treated with dichloromethane (50 ml) and an 10% NaOH aqueous solution (30 ml). The organic phase was dried over Na2SO4 and evaporated to get 38 (Figure 23) as a colourless oil (1.48 g, 61%); 1H NMR δ 1.31 (d, J = 6.4 Hz, 6H), 2.10 (s, 3H), 2.68–2.82 (series of m, 4H), 3.79 (s, 3H), 4.44 (sept, J = 6.4 Hz, 1H), 6.66 (dd, J = 2, 8 Hz, Ar, 1H), 6.68 (d, J = 2 Hz, Ar, 1H), 6.77 (d, J = 8.0 Hz, Ar, 1H); 13C NMR δ 22.10, 29.36, 30.04, 45.35, 55.86, 71.51, 112.36, 116.24, 120.06, 134.13, 145.53, 146.16, 150.35, 208.11; Anal. Calcd. for C14H20O3: C, 71.16; H, 8.53; Found: C, 71.20; H, 8.56.
Evaluation of antioxidant potential (pro-oxidant/antioxidant activity) of the 36 compounds
Healthy volunteers who had attended their regular medical check-up at the Military Medical Academy in Belgrade and had given approval that any serum left over after biochemical analyses planned by physicians could be used for this study. Fifty samples whose basic biochemical parameters were within metabolite reference ranges were selected. After thorough mixing the serum pool was aliquoted into 450 µl portions and frozen at –83°C until analyses took place.
The stock concentration of each of the compounds was 10 mg/ml. DMSO was the solvent. All analyses were performed in triplicate. To 450 µl of serum 50 µl of each compound under investigation was added (500 µl total) and then incubated at 37°C for 2 h.
The same procedure was implemented for the samples with concomitant presence of tested substances and tert-butyl-hydroperoxide (TBH) (0.5 µl/ml solution in distillate water) as the pro-oxidant substance.
Total oxidative potency (TOP)
TOP was determined according to Erel [33] and Kotur-Stevuljevic et al. [34]. All oxidants in the sample (for example H2O2 and lipid hydroperoxides) oxidise a ferro-orthodianisidine complex to ferric ion in an acidic environment in the presence of glycerol. The resulting ferric ion forms a coloured complex with xylene orange. Colour intensity is measured spectrophotometrically (A 560 nm) and is proportional to the total content of oxidising molecules in the sample. The assay is calibrated with H2O2 (10–200 µmol/l) and the results were expressed in µmol H2O2 Equiv/l.
Pro-oxidant-antioxidant balance (PAB)
The PAB indicates concomitant pro-oxidant load and antioxidative capacity of a particular organism. A modified version of a published method [35] was used for PAB determination. The assay determines the concentration of H2O2. The chromogen 3,3’,5,5’-tetramethylbenzidine (TMB) reacts with both H2O2 and antioxidants (including uric acid and other reducing species). The reaction between H2O2 and chromogen is catalysed by the enzyme peroxidase, resulting in oxidation of TMB to produce an intense colour. In contrast, the reaction between uric acid and similar compounds with chromogen is not catalysed by peroxidase, causing discolouration. The colour generated in the reaction is proportional to the ratio of pro-oxidants and antioxidants. Absorbance was read at 450 nm after a 10 min incubation of the reaction medium at 37°C.
SH-groups (SHG)
Total sulphydryl groups in serum was determined by a modification of Ellman’s method [36] (according to Kotur-Stevuljevic et al.) [34], based on the formation of a yellow-coloured reaction product between 2,2’-dinitro-5,5’-dithiobenzoic acid (DTNB) and aliphatic thiol compounds in basic conditions (pH = 9.0). Absorbance was measured at 412 nm.
Total antioxidant capacity (TAC)
TAC was measured using the stable ABTS+ cation as a chromogen [37]. ABTS is oxidised by H2O2 in acetate buffer; pH = 3.6 to a green coloured ABTS+ cation. Antioxidants present in the sample lead to varying degrees of discoloration proportional to their concentration (the antioxidant potential of the sample). After incubation for 10 min at room temperature absorbance at 600 nm was recorded.
Pro-oxidative Score, Antioxidative Score and Oxy Score
Overall oxidative stress was evaluated through several scores: Antioxidative Score, Pro-oxidative Score and Oxy Score as previously suggested [38]. The Antioxidative Score (indicating protective capacity) was calculated as the mean of the Z scores of the measured antioxidant parameters – TAC and SHG. The Pro-oxidative Score (indicating damage potential) was calculated as mean Z scores of the measured pro-oxidant parameters – TOP and PAB. The Oxy Score was calculated as the difference between Pro-oxidative and Antioxidative Scores. Population parameters for calculation of Z scores were from our previous investigations using a healthy population. The formula for Z score calculation was as follows: (xi – x)/σ, where xi is sample value, x is population mean, and σ is population standard deviation. A higher Oxy Score indicates less antioxidative protection, in other words a more pronounced pro-oxidative state.
Statistical analysis
The results of all parameters were expressed as percentiles (medians with 25th and 75th percentile values). The non-parametric Kruskal-Wallis test with post hoc Mann-Whitney U test with Bonferroni correction was used for statistical analysis. Values of p < 0.05 were considered statistically significant.
Results
The material for antioxidant potential assays comprises 36 compounds (monomers or their C2-symmetric dimers) whose synthesis started, for almost all of them, from commercially available, naturally occurring compounds (Table I) and following methods known in the literature or improving them with sustainable reagents and procedures. Compound 1 was prepared by condensation of 5,6-dimethoxybenzofuran-3(2H)-one with 4-methoxy-benzaldehyde in the presence of neutral alumina (with good yield). The same procedure was followed for the preparation of dimer 2, starting from the corresponding benzofuran-3-one dimer. Compound 5 was prepared in one pot (in 60% yield) by a coupling reaction of 4-(4-isopropoxy-3-methoxyphenyl)butan-2-one in the presence of molybdenum (V) chloride. The coupling reaction of monomer 10 in the presence of methyl-tributylammonium permanganate at room temperature produced dimer 11 (in 61% yield). Claisen-Schmidt condensation of 5,5’-diethoxy-6,6’-dihydroxy-[1,1’-biphenyl]-3,3’-dicarbaldehyde with acetone under basic conditions gave compound 13 (in 80% yield). Following the same reaction conditions, compound 21 was prepared starting from per-O-acetylated β-C-glucopyranosyl ketone and 5,5’,6,6’-tetramethoxy-[1,1’-biphenyl]-3,3’-dicarbaldehyde and further deacetylation (with 73% overall yield). Following the same procedure, compound 20 was prepared (in 80% yield) starting from veratraldehyde. Compounds 14, 15, 17, 18, 19 and 40 were obtained by protection of the corresponding phenolic hydroxyl group with the appropriate organohalide under basic conditions, in acetone at reflux. The coupling reaction of raspberry ketone 22 in the presence of methyl-tributylammonium permanganate as a catalyst gave dimer 23. Microwave procedures were applied for the preparation of compound dimers 25–29, 31 and monomer 26 following different reaction conditions. Dimer 25 was prepared (in 60% yield) starting from the commercial 4,4’-dihydroxy biphenyl and hexamethylenetetramine in the presence of trifluoroacetic acid. A palladium catalysed Heck reaction between 3-methylpent-1-en-3-ol and 2-bromophenol under basic conditions gave compound 26. Following the same procedure for 26 and starting from the corresponding dimer dibromide, compound 27 was obtained (in 40% yield). Apocynin and 4,4’-bis(allyloxy)-[1,1’-biphenyl]-3,3’-dicarbaldehyde in the presence of lithium hydroxide in methanol produced dimer 29 (in 40% yield) after 30 min of microwave irradiation. Following the same procedure of 29, compound 31 was obtained (in 31% yield) starting from 4,4’-bis(allyloxy)-[1,1’-biphenyl]-3,3’-dicarbaldehyde and 3,4-dimethoxyacetophenone. Monomer 30 was prepared (in 62% yield) following Claisen-Schmidt condensation of 3,4-dimethoxy-acetophenone and 2-(allyloxy)benzaldehyde under basic conditions.
Based on the values of the calculated Prooxidative Score and the calculated Antioxidative Score expressed together as the Oxy Score (Table I), six compounds (6, 7, 12, 13, 26, and 27) were found to have strong antioxidant properties, and four compounds (2, 29, 30, and 31) were found to have extremely weak antioxidant properties in the serum pool assay. Table II shows the results of all the distinct redox status markers measured in our current study using the in vitro assays.
Table II
Redox status parameters measured after the reaction of polyphenolic compounds with serum
The group of weak antioxidants (compounds 2, 29–31) caused a significant increase in both PAB and TOP parameters compared to the group of strong antioxidants (compounds 6, 7, 12, 13, 26 and 27). Strong antioxidants significantly increased TAC values compared to weak antioxidants. After addition of exogenous pro-oxidant TBH in samples containing strong antioxidants, we noted a decrease in TOP concentration for compounds 6, 12 and 13, whereas for compounds 7 and 27 TOP concentration was unaffected. Compound 26 unexpectedly increased TOP. Because of obvious interactions between Ellman’s reagent and polyphenolic compounds we omitted SHG from the calculation of the Antioxidative Score. When comparing the same parameters in the weak antioxidant compound group before and after TBH addition we found that PAB and TOP both diminished. All differences were significant (p < 0.05). Unexpectedly TAC increased after TBH addition in the weak antioxidative compound group (p < 0.05).
Regarding the influence of TBH addition in samples of the strong antioxidant group we noted a significant increase in PAB values only for compound 26 (p < 0.05). The PAB parameter remained at the basic level (without TBH) for the other five strong antioxidants.
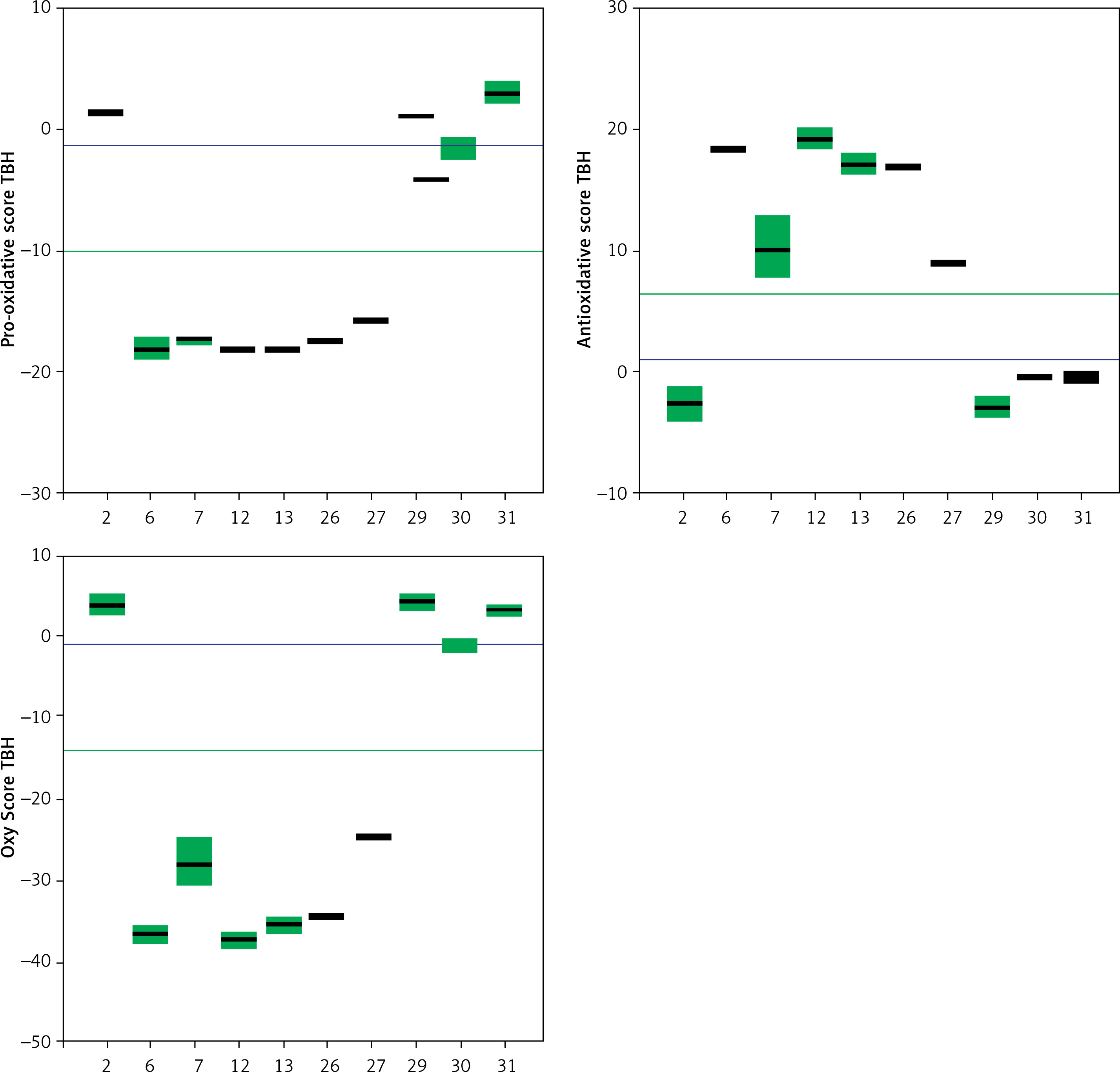
Trolox, a known and well-characterized antioxidative substance, was used as a control. In our assay its antioxidative activity was found to lie between that of the weak and strong groups of compounds. The Oxy Score values of the strong antioxidative compounds ranged from –28.8 ±0.4 to –32.5 ±1.1, while the Oxy Score values of weaker antioxidative compounds were significantly more positive and ranged from 11 ±0.7 to 14.6 ±0.8. The Oxy Score for Trolox was –7.01 (–7.53– –6.51), so it was clearly positioned between strong and weak antioxidative compounds. There was a statistically significant difference in the Oxy Score between all strong antioxidative compounds and all weak antioxidative compounds (p < 0.001). This was also true for strong antioxidative compounds and Trolox and weak antioxidative compounds and Trolox (both p < 0.01). Figures 24 and 25 show the results of the 6 strongest antioxidative compounds and 4 weakest antioxidative compounds. The green line presents the Trolox value. Using the non-parametric Kruskal-Wallis and post hoc Mann-Whitney U test it was found that there was no statistically significant difference (p > 0.05) in the values of the Oxy Score (antioxidant activity capacity) of the 6 strongest antioxidative compounds (in assays without TBH (Figure 24) and in those with TBH added (Figure 25)).
Figure 24
Values of Pro-oxidative, Antioxidative, and Oxy Scores of six strong (compounds 6, 7, 12, 13, 26, 27) and four weak (compounds 2, 29, 30, 31) antioxidants in the serum matrix without TBH addition. The green solid line represents the value obtained with Trolox (standard), the blue solid line represents values obtained with serum with DMSO as solvent (control)
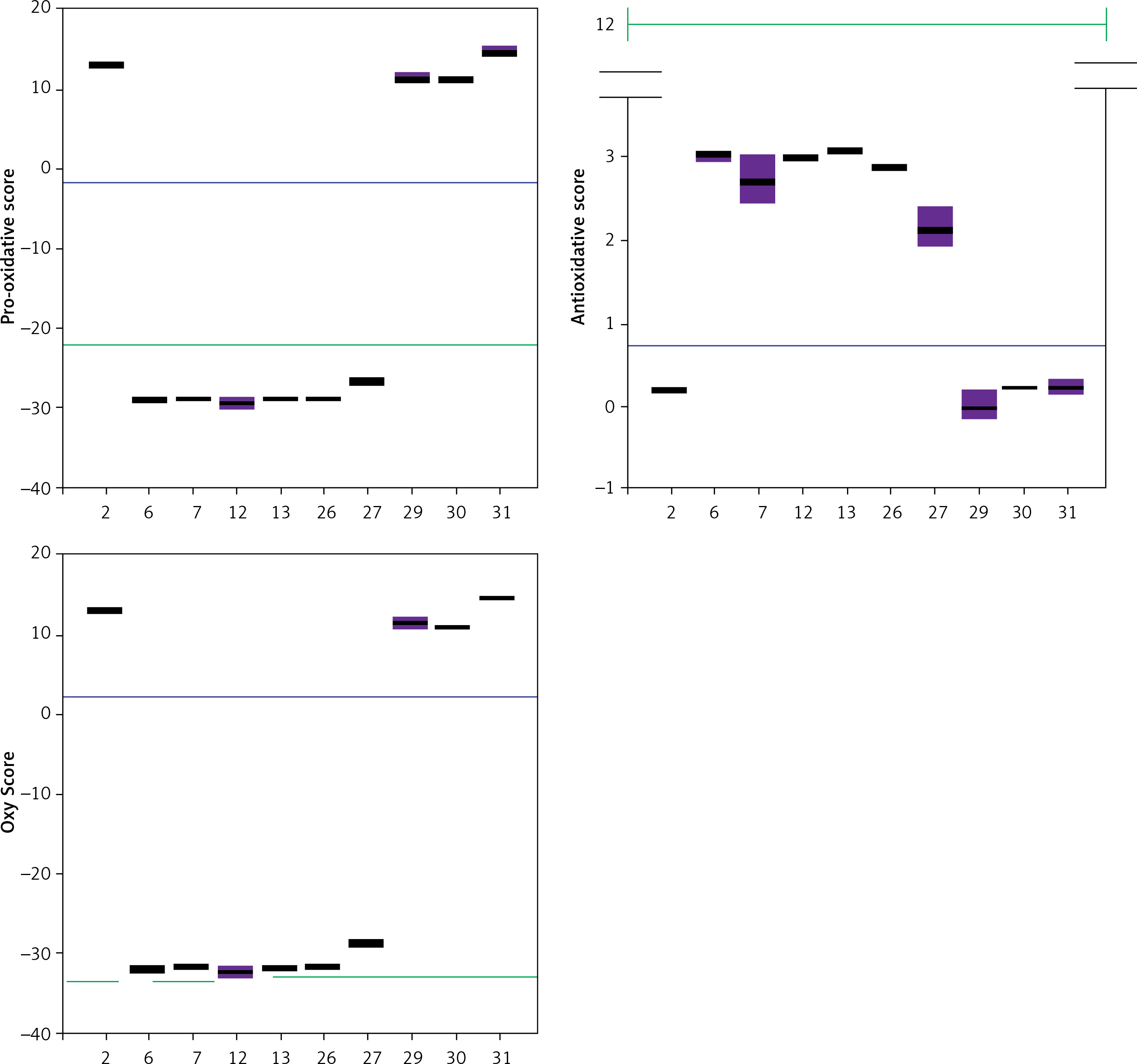
Figure 25
Values of Pro-oxidative, Antioxidative, and Oxy Scores of six strong (compounds 6, 7, 12, 13, 26, 27) and four weak (compounds 2, 29, 30, 31) anti-oxidants in the serum matrix with TBH addition. The green solid line represents the value obtained with Trolox (standard). The blue solid line represents values obtained with serum with DMSO as solvent (control)
Lipophilicity (logP) of the strongest antioxidative compounds in serum (compounds 6, 7, 12, 13 and 26) varied between 1.27 and 2.85 whereas higher logP-value were calculated for the weakest antioxidative compounds (compounds 2, 29, 30, 31) in the range of 3.90–7.44.
A similar trend was calculated in the series of antioxidants and pro-oxidants assayed without addition of human serum (Table I). Trolox’s logP value (3.19) was close to the series of compounds with the strongest antioxidative activity.
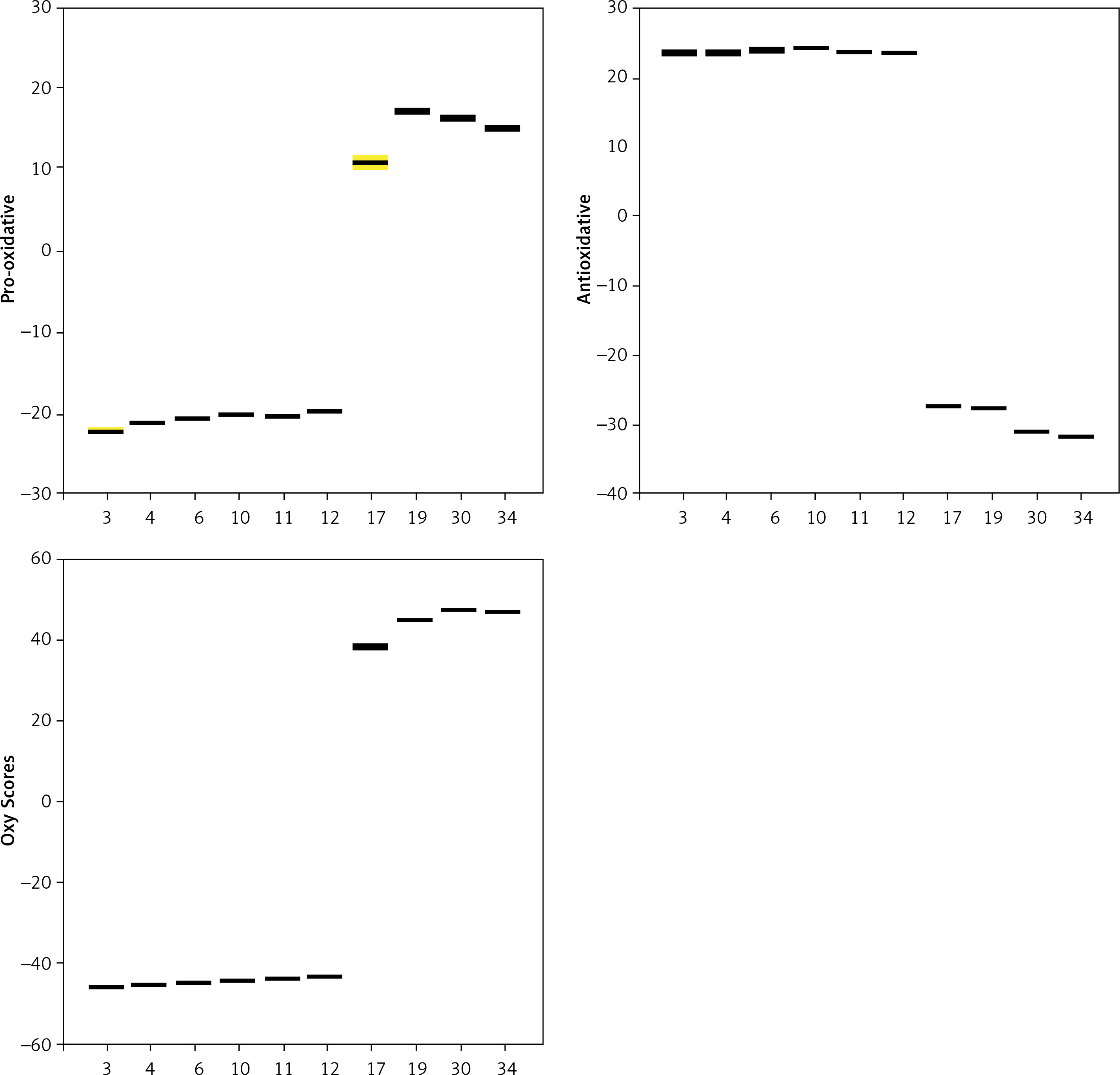
Results of in vitro analysis in samples in non-biological matrix (direct reaction of selected polyphenolic compounds with reagents for TAC, TOP and PAB determination, without addition of human serum) show a statistically significant difference between the parameters determined (p < 0.001). Compounds 3, 4, 6, 10, 11 and 12 were found to be the most effective antioxidants, while compounds with the weakest antioxidant activity were 17, 19, 30 and 34 (Figure 26). There was no statistically significant difference in the group of the six newly selected potent antioxidants (p > 0.05). Figure 26 shows the obtained values of the six strongest antioxidative compounds and four weakest antioxidative compounds in analysis in samples in the absence of serum.
Figure 26
Values of Pro-oxidative, Antioxidative, and Oxy Scores of the 6 strongest antioxidative compounds (3, 4, 6, 11, 12) and the 4 weakest antioxidative compounds (17, 19, 30, 34) in samples without addition of human serum
Our results so far have identified the 6 strongest antioxidative compounds and the 4 weakest antioxidative compounds. The strongest were dehydrozingerone derivates, monomers or dimers (compound 6 and its structural analogue compound 12 (monomer), compounds 7 and 13 (dimers)), prenylated phenol and 4,4’-dihydroxybiphenyl derivative (compounds 26 (monomer) and 27 (dimer)). The weakest were aurone and chalcone derivatives (compounds 2, 29 and 31 (dimers)) and one chalcone monomer (compound 30).
The strongest in vitro antioxidant activity in the matrix in the absence of human serum was that of zingerone (compounds 3 and 4, monomer and dimer, respectively), dehydrozingerone (compound 6), an ethyl vanillin 3-buten-2-one derivative monomer (compound 12) and the vanillyl alcohol methyl esters monomer (compound 10) and dimer (compound 11). The weakest antioxidants were the dehydrozingerone dimer derivative (compound 17), a dimer of ethyl vanillin 3-buten-2-one derivative (compound 19), a methylmagnolol derivative (compound 34) and a chalcone derivative (compound 30).
It is important to note that compounds 6 and 12 (strong antioxidative activity) and compound 30 (weak antioxidant activity) manifested equal activity both in the absence and presence of human serum.
Discussion
The pro-oxidant/antioxidant activity of polyphenolic compounds is dependent on various factors including metal-reducing potential, chelation properties, pH, solubility and concentration [39]. Such factors together with polyphenolic compound bioavailability and stability in biological environments need be considered when evaluating their potential antioxidant bioactivity [17, 40]. There have been many polyphenolic compounds investigated so far for potential use in human medicine, but resveratrol is especially appreciated primarily for its antioxidant and anti-inflammatory potency. However, Chudzińska et al. concluded that its beneficial effects are still not clearly confirmed and that there is a need for further and better controlled clinical studies [41]. On the other hand, Patti et al. [42] confirmed beneficial effects of olive oil, as a phenolic compound, on anthropometric and biochemical parameters, including inflammatory markers in patients with metabolic syndrome and hepatic steatosis.
Molecules having two symmetric binding moieties bearing a flexible bridge of suitable length and properties would be expected to show a higher target binding affinity leading to higher biological activity compared with molecules where one binding moiety is missing [21, 43, 44].
Due to their specific structure, hydroxylated biphenyls have been found to have better antioxidant effects than the corresponding monophenols [17, 39, 45]. The results obtained in our study confirm the finding that the strongest antioxidant effect in the (serum-containing) biological environment was achieved by compounds containing a free phenolic hydroxyl as an electron-donating group with an ethoxy/methoxy in ortho position and the side chain with an α,β-unsaturated ketone, which contributes to the stability of phenoxyl radicals by increasing electronic delocalisation in the bio-environment [18].
But interestingly, our results showed that a dehydrozingerone monomer (compound 6) and ethyl vanillin 3-buten-2-one derivative (compound 12) exhibited better antioxidant effect than their corresponding dimers, even though an almost comparable Oxy Score was calculated for the corresponding dimers (compounds 7 and 13, respectively). The difference in the antioxidant activity of the dimers could have been due to the formation of intramolecular hydrogen bonds and reduced rotation (appearance of axial chirality) that hinders electronic delocalisation of the phenoxyl radical through the two non-planar aromatic rings [46].
Members of the vitamin E family, known as tocopherols, are methyl-substituted tocol derivatives. The most potent is α-tocopherol (5,7,8-trimethyltocol) which contains three electron-donating methyl groups that increase the nucleophilicity and reactivity of the phenolic group at position 6 of the chromane ring. Additionally, resonance stabilization by the para oxygen in the chromane ring improves the stability of the α-tocopherol radical [47].
One of the primary functions of vitamin E is preventing the oxidation of lipids, particularly unsaturated fatty acids, through its antioxidant effects. Vitamin E is a fat-soluble compound and penetrates biological membranes. Located in cell membranes, vitamin E could act toward reactive oxygen species (ROS), thus protecting cellular components from oxidative damage [47]. α-Tocopherol donates its electrons to the free radicals to neutralize them. In this process, α-tocopherol is fully oxidized to the α-tocoquinone form and loses its antioxidant capacity.
Trolox is a water soluble analogue of α-tocopherol, used as a reference in the evaluation of antioxidant activities of compounds [48]. In this work, Trolox was used as a positive control.
Lipophilicity of Trolox and compounds 1-36 was estimated by ChemBioDraw Ultra 13.0 software (CambridgeSoft) expressed as logP (the logarithm of the partition coefficient for n-octanol/water) and listed in Table I. Although a carboxylic group is present in the structure of Trolox, the high logP value of the compound (3.19) implies a lipophilic character, as already documented in another study [20]. The physicochemical properties of Trolox would explain the high affinity of the compound towards cellular membranes [48], but also could account for Trolox’s affinity towards lipoprotein particles, i.e. their lipid content. We could hypothesise a different partition of Trolox in the bilayer surface that influences the interactions of the compound with peroxyl radicals generated in a selected experimental method. This point offers an explanation for the medium antioxidative Trolox activity positioned between strong and weak polyphenolic compounds analysed in this study.
An unexpectedly strong antioxidant effect was exhibited by compound 26, which lacks a guaiacyl unit and an α,β-unsaturated methyl ketone side chain as in compounds 6 and 12. It is possible that both in serum and under acidic or basic conditions, compound 26 could undergo elimination of water by proton removal from the methylene group of the allylic chain and subsequent formation of a prenylated unit that would stabilise the phenoxyl radical. Good antioxidant activity of this structural moiety was also confirmed in the corresponding dimer, compound 27.
Although there have been many studies in animal models using zingerone as an antioxidant and anti-inflammatory compound [49], our results indicate that zingerone (compound 3) exhibited greater antioxidant activity in an assay without biological material (human serum).
Zingerone exhibits good radical-scavenging activity but poorly acts as a chain-breaking antioxidant in lipid auto-oxidation in comparison with dehydrozingerone 6 and its corresponding dimer 7. Lipophilic antioxidants play a crucial role in attenuating oxidative processes that occur in cell membranes in diseases including cancer and neurodegeneration [49–51].
We found that lipophilicity of the strongest antioxidants detected in human serum was lower in comparison to that of the weakest antioxidants which contained a protected alkyl phenolic-hydroxyl group. It is possible that hydrolytic enzymes present in human serum are not able to deprotect the alkylated phenolic-hydroxyl group that could occur in another biological system [52, 53].
Raspberry ketone, compound 22, one of the main components of red berry fruits and responsible for inhibiting inflammatory processes [43, 54], exhibited a modest Oxy Score. The structure of compound 22, lacking a methoxyl group in ortho position to the phenolic-hydroxyl group and an α,β-unsaturated lateral chain, did not achieve a calculated Oxy Score value as great as dehydrozingerone 6. Nevertheless, raspberry ketone has been considered a health-promoting compound and marked as a food supplement [55].
Chalcones are a group of polyphenolic compounds that have recently been used as additives and ingredients in cosmetic preparations because of their potential high antioxidant and anti-inflammatory effects [56, 57]. However, the tested compounds in our study, which are monomers and dimers of chalcones and aurones (formed by cyclisation of chalcones), namely compounds 2, 29, 30 and 31, exhibited the weakest antioxidant properties. The reason for this is probably the small number and/or absence of free phenolic hydroxyl groups in the molecule (which are mandatory for the antioxidant action of polyphenols), their too high lipophilicity or their inadequate configuration.
The part of results which showed comparison before and after TBH addition and opposite activity in a group of weak compared to strong antioxidants could be explained by the lack of any prooxidative capacity in weak antioxidants, which could, according to Halliwell, initiate triggering of a fast and strong antioxidative response of biological medium [58, 59].
In conclusion, from a diverse group of tested natural-like polyphenolic compounds comprising zingerone, dehydrozingerone, aurone, chalcone, magnolol derivatives, monomers and their corresponding dimers, the greatest antioxidant activity was that of dehydrozingerone analogues (compounds 6 and 12). In the future we will focus on these two moieties to confirm their mechanism of antioxidant action. The results suggest that those compounds could be candidates for the curcumin analogues that potentially improve their bioavailability in vivo. The most convincing confirmation of their antioxidative potency also comes from the results of the same activity achieved by the well-known antioxidant Trolox, which is a water-soluble vitamin E analogue. Trolox’s activity was found between the strong and weak antioxidant compounds analysed in our study. This means that compounds 6 and 12 are worthy of further investigation into the antioxidant biology.
The inconsistency of our results regarding the antioxidant effects of the compounds in different matrices (human serum and in vitro environment without bio-matrix) supports the unpredictability of polyphenolic pharmacophore behaviour in a biological environment and the difficulty of elucidating the presumed mechanism of action. Selected dehydrozingerones would be good candidates for in-depth testing of their biological behaviour and for possible precursors for the synthesis of novel polyphenolic molecules for potential therapeutic applications. More studies (animal and human or cell culture based) are necessary to provide evidence and to elucidate dose-response and cost-benefit relationships between polyphenol-like compounds, their therapeutic potential and health benefits. One of the limitation of this current study is the acellular material used (human serum), but this is available biomaterial upon which we have developed a platform for testing different substances’ interaction with biomolecules of human origin.