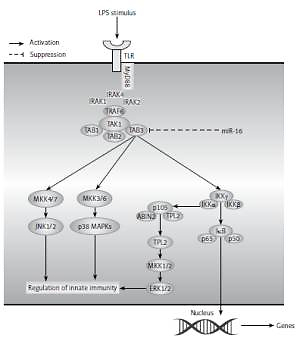
Introduction
Intervertebral disc degeneration (IDD) is identified as a key facilitator of low back pain. However, the occurrence of IDD disease has remained unclear due to being involved in complex biochemical and cytological processes. Previous studies have indicated that imbalance of anabolic and catabolic gene expression in chondrocytes of the nucleus pulposus (NP) can result in the occurrence of IDD [1]. Through upregulation of some molecules such as pro-inflammatory cytokines and extracellular matrix (ECM) degrading enzymes, homeostasis of catabolism and anabolism in the ECM of the intervertebral disc (IVD) shifts toward a degenerative and catabolic state with subsequent breakdown of ECM components, including collagen and proteoglycan of proteoglycans (PGs) [2, 3]. Lipopolysaccharide (LPS) was proved to be an inflammatory factor with strong potency to induce inflammatory responses of human cells. Furthermore, LPS could downregulate the production of collagen and PGs in NP cells and induce a variety of proinflammatory cytokines and matrix-degrading enzymes (including MMP-3, MMP-13, ADAMTS4 and ADAMTS5), causing a decrease of PGs’ content and the occurrence of IDD. As a TLR (Toll-like receptor) ligand, LPS initiates TLR signal transduction by the TLR4 receptor to increase the expression of proinflammatory cytokines (TNF-α, interleukin (IL)-1β, IL-6) and MMPs. Proinflammatory cytokines, such as interleukin-1β (IL-1β) and tissue necrosis factor-α (TNF-α), can also facilitate the development of inflammation in IDD progression, and subsequently accelerate the occurrence of IDD. Nuclear factor κB (NF-κB) is a ubiquitous transcription factor. It can be induced by a plethora of stimuli and further translocates into the nucleus to mediate expression of numerous genes, such as cytokine genes and enzymatic genes. In the TLR signaling pathway, the stimulation of TLR initiates signaling cascades, and further leads to activation of transcription factors NF-κB and kinases 5, triggering coding immunity and an inflammatory response [4]. Therefore, NF-κB plays a pivotal role in the regulation of IDD progression.
MicroRNAs (miRNAs) are a type of small single-stranded noncoding RNA molecule 18–22 nt in length. MiRNAs can bind to the seed sequence of the 3′-untranslated region (3′-UTR) of its target mRNA to repress gene expression by inducing mRNA degradation or translation repression [5, 6]. More importantly, miRNAs perform complex functions in most cells, mainly including cell growth, proliferation and differentiation, signal transduction, apoptosis, metabolism and aging. Increasing evidence has demonstrated that miRNAs have a pivotal position in the immune system, such as immunomodulation, development and differentiation of B and T cells, proliferation of monocytes and neutrophils, transition of antibodies and release of inflammatory factors [7, 8]. In many immune cells (monocytes, macrophages, etc.), the expression of many miRNAs will change after LPS induction, including miR-146, miR-155 and the let-7 family, which have been reported already. These miRNAs positively or negatively regulate TLR signals by targeting components in the TLR pathway [9–11]. For example, miR-146 can downregulate the expression of TRAF6, TAB2 and IRAK2. In addition, the role of many miRNAs in the pathogenesis of IVD has been reported [12]. However, the function of miR-16 in IDD remains unclear. Previous studies indicated that miR-15a and miR-16 presented increased expression in the serum of newborns with sepsis, and inhibited LPS-induced inflammatory pathways [13]. Pauley et al. found abnormal expression of miR-16 in synoviocytes, tissues and peripheral blood of patients with rheumatoid arthritis, which implied that miR-16 is relevant to immunoregulation [14]. Based on the above findings, we hypothesized that miR-16 may exert a protective role in the pathogenesis of IDD. The current study tested the role of miR-16 in regulating the LPS-induced inflammatory response of NP cells, and further investigated the regulatory mechanism.
Material and methods
Cell extraction and culture
Sprague-Dawley (SD) rats aged 6 weeks were purchased from the Experimental Animal Center of Shanghai (Shanghai, China). Animal experiments were approved by the Ethics Committee of The First Affiliated Hospital, Jinan University. Firstly, Sprague-Dawley (SD) rats aged 1 month were collected to obtain the whole lumbar vertebra under sterile conditions. Jelly-like NP cells were extracted from the lumbar vertebra under a microscope and centrifuged for 5 min at 1000 r/min, then digested with 2.5 g/l trypsin (Sigma-Aldrich, St. Louis, MO, USA) and 1 g/l collagenase II (Sigma-Aldrich, St. Louis, MO, USA) in turn. Finally, the NP cells were cultured to passage 2–3 by DMEM complete medium supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO, USA), 100 U/ml penicillin (Sigma-Aldrich, St. Louis, MO, USA) and 100 mg/ml streptomycin (Sigma-Aldrich, St. Louis, MO, USA).
RNA isolation and quantitative real-time reverse transcriptase PCR (qRT-PCR)
Total RNA of NP cells was extracted using TRIzol regent (Invitrogen Inc., Carlsbad, CA, USA) and miRcute miRNA isolation kits (Tiangen Biotech, Beijing, China). Then, the total RNA was reverse transcribed into first strand cDNA using the miRcute miRNA First-Strand cDNA Synthesis kit (Tiangen Biotech, Beijing, China). q-PCR was performed on the ABI PRISM 7900 system (Applied Biosystems, Grand Island, NY, USA). Each PCR reaction contained 10 μl of 2 × SYBR Green Real-time PCR Master Mix plus 2 μl of Plus Solution, 1 μl of each primer (10 μM), 2 μl of cDNA template, and 4 μl of ddH2O in a 20 μl reaction volume. The temperature profile for gene expression was 95°C for 60 s followed by 40 cycles of amplification (95°C for 15 s, 60°C for 15 s, and 72°C for 40 s). The q-PCR reaction conditions for miRNA-16 detection were as follows: pre-denaturation at 95°C for 10 min; denaturation at 95°C for 15 s, annealing at 60°C for 1 min and extension at 72°C for 2 min (40 cycles); and a final extension step at 72°C for 7 min. Gene expression level was calculated by the 2–ΔΔt method. β-actin and U6 served as internal references. The primer sequences for the genes are available in Table I.
Table I
Primers for the clone and RT-PCR of genes
Cell transfection
Firstly, the open reading frame (ORF) of the TAB3 gene was amplified with cDNA of NP cells as a template, and then connected with linearized pcDNA3 to construct the recombinant plasmid pcDNA3-TAB3. Following that, the pcDNA3-TAB3 was amplified. The amplified fragment was combined with the eukaryotic expression vector pEGFP to construct the recombinant plasmid pEGFP-TAB3 (Clontech Laboratories, Inc., Mountain View, CA, USA). MiR-16 mimics and the miR-16 inhibitor were synthesized by Shanghai Sangon Biological Engineering Technology & Services Co., Ltd. (Shanghai, China). NP cells were transfected with miR-16 mimics, the miR-16 inhibitor and TAB3 over-expressing plasmids. The co-transfection was conducted in NP cells with miR-16 mimics and TAB3 over-expressing plasmids. Liposome 2000 (Invitrogen Inc., Carlsbad, CA, USA) was used for transient transfection of NP cells. After cell transfection, NP cells were stimulated with LPS (10 μg/ml) (Sigma-Aldrich, St. Louis, MO, USA) for various lengths of time.
ELISA determination
Expression levels of ECM degrading enzymes and NO-related genes were measured using an ELISA regent kit (R&D Systems, Minneapolis, MN, USA). The experiment was carried out strictly according to the instructions.
Western blotting
The obtained NP cells were washed 2–3 times with cold phosphate buffered saline (PBS) (Sigma-Aldrich, St. Louis, MO, USA), and then lysed with RIPA lysis buffer (Sigma-Aldrich, St. Louis, MO, USA) to extract total protein. The concentration of the total protein was determined using a BCA kit (Thermo Fisher Scientific, San Jose, CA, US). A volume of 20 mg of protein sample was drawn to the gel, separated by 10% SDS-PAGE, then transferred to polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Bedford, MA, USA). The transfer membrane was sealed for 1 h with 5% skim milk at room temperature, washed once with PBS, and incubated overnight with primary antibodies at 4°C. The primary antibodies were as follows: anti-Aggrecan (ab36861, 1 : 200, Abcam, Cambridge, MA, USA), anti-Collagen II (ab185430, 1 : 200, Abcam, Cambridge, MA, USA), anti-TAB3 (ab85655, 1 : 2000, Abcam, Cambridge, MA, USA), and anti-GAPDH (ab9484, 1 : 1000, Abcam, Cambridge, MA, USA). Following that, the transfer membrane was washed 3 times with TBST (TBS with Tween 20) (Sigma-Aldrich, St. Louis, MO, USA). The secondary antibody combined with HRP (ab131368, 1 : 1000, Abcam, Cambridge, MA, USA) was used for incubation with the transfer membrane for 1 h. Protein bands were detected with an Enhanced Chemiluminescence assay kit (Thermo Fisher Scientific, San Jose, CA, USA). Images were captured using a ChemiDoc XRS + image analyzer (Bio-Rad Laboratories, Inc. CA, USA). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody served as the internal reference.
Luciferase assay
TargetScanHuman 7.0 (http://www.targetscan.org/) and miRBase (http://www.mirbase.org/) microRNA databases were used to identify the target genes of miR-16. The primers of TAB3 3′-UTR and mutant TAB3 3′-UTR that may contain binding sites of miR-16 were designed and synthesized. The two kinds of target fragment were cloned into the pmirGLO dual Luciferase Reporter (Promega Corp., Madison, Wisconsin, USA) to construct the wild-type pmirGLO-TAB3 vector and mutant pmirGLO-TAB3 vector. Following that, the NP cells were transfected with the two types of vectors and miR-16 mimics or negative controls. After 24 h of transfection, the cells were collected. The luciferase was detected by the Luciferase reporter assay reagents (Promega Corp., Madison, Wisconsin, USA).
Determination of NF-κB activity
One day before transfection, the NP cells were placed into a 96-well plate at 5 × 103 cells/well. Co-transfection was performed using lipofectamine 2000 (Invitrogen Inc., Carlsbad, CA, USA) with 20 ng of NF-κB luciferase reporter and 5 ng of pRL-TK vector in each well (Promega Corp., Madison, Wisconsin, USA). After that, the cells were cultured for 24 h and then collected. The luciferase was detected by the Dual Luciferase Reporter Assay System (Promega Corp., Madison, Wisconsin, USA).
Statistical analysis
Data are expressed as the mean ± standard deviation. All data were processed using SPSS 20.0 software (SPSS, Inc., Chicago, IL, USA). One-way analysis of variance was performed for multiple comparisons and differences between two groups were compared using unpaired Student’s t-test for parametric data or the Mann-Whitney rank sum test for nonparametric data. P < 0.05 was considered to indicate a statistically significant difference.
Results
MiR-16 was downregulated in NP cells after LPS induction
A concentration of 10 μg/ml LPS was used to induce the NP cells. Compared with the control group, the gene expression of miR-16 in the NP cells was significantly decreased after 12 h of induction, and decreased still further after 24 h and 48 h (p < 0.001) (Figure 1 A).
Figure 1
A – Gene expression of miR-16 was suppressed by LPS treatment in NP cells (p < 0.01). B – MiR-16 stimulated the mRNA expression of aggrecan and collagen II (p < 0.01). C – MiR-16 stimulated the protein expression of aggrecan and collagen II (p < 0.01). D – MiR-16 suppressed the expression of MMP3 and MMP13 induced by LPS (p < 0.01, p < 0.001). E – MiR-16 suppressed the expression of ADAMTS4 and ADAMTS5 induced by LPS (p < 0.01, p < 0.001)
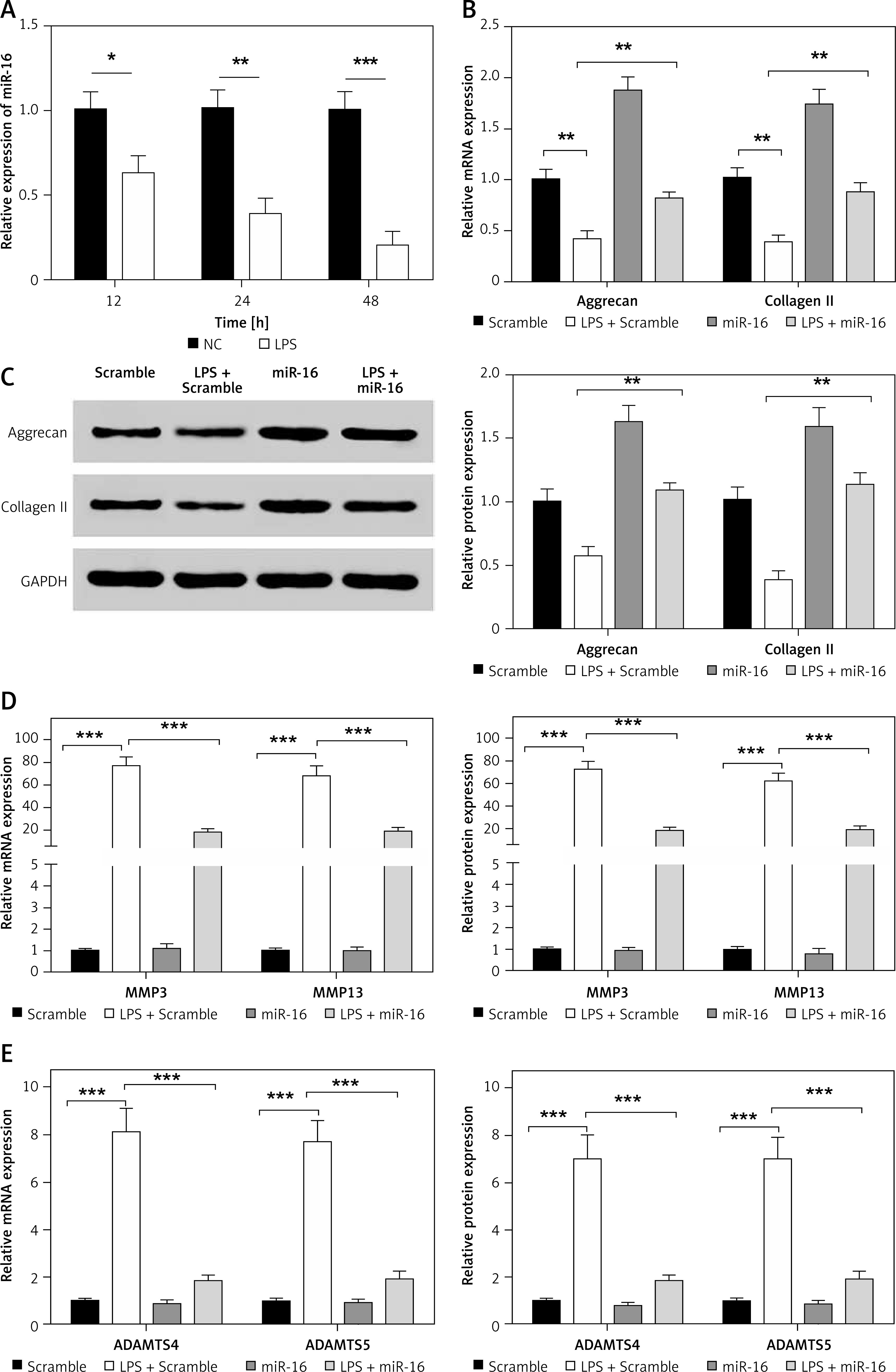
Overexpressed miR-16 regulated the expression of ECM genes and synthesis of ECM degrading enzymes
The results indicated that LPS significantly downregulated the expression of aggrecan and collagen II in NP cells (Figures 1 B, C). The expression of aggrecan and collagen II in miR-16 mimics transfected NP cells was significantly higher than that in the Scramble group (used as control group) before LPS induction (p < 0.05), and significantly higher than that in the LPS + Scramble group after LPS induction (p < 0.01). The protein expression of aggrecan and collagen II was consistent with their mRNA expression after LPS induction.
In order to confirm the regulatory effect of miR-16 on the expression of ECM degrading enzymes (MMP3, MMP13, ADAMTS4, ADAMTS5) in NP cells, a qRT-PCR experiment was carried out to determine their expression levels. The results showed that the gene expression of these degrading enzymes was significantly increased in the LPS + Scramble group compared with the Scramble group (p < 0.001), while transfection of miR-16 suppressed the up-regulation of LPS induced ECM degrading enzymes (p < 0.001). Additionally, the results of ELISA revealed that overexpressed miR-16 can significantly inhibit the protein expression of these degrading enzymes in LPS-induced NP cells (p < 0.01, p < 0.001) (Figures 1 D, E).
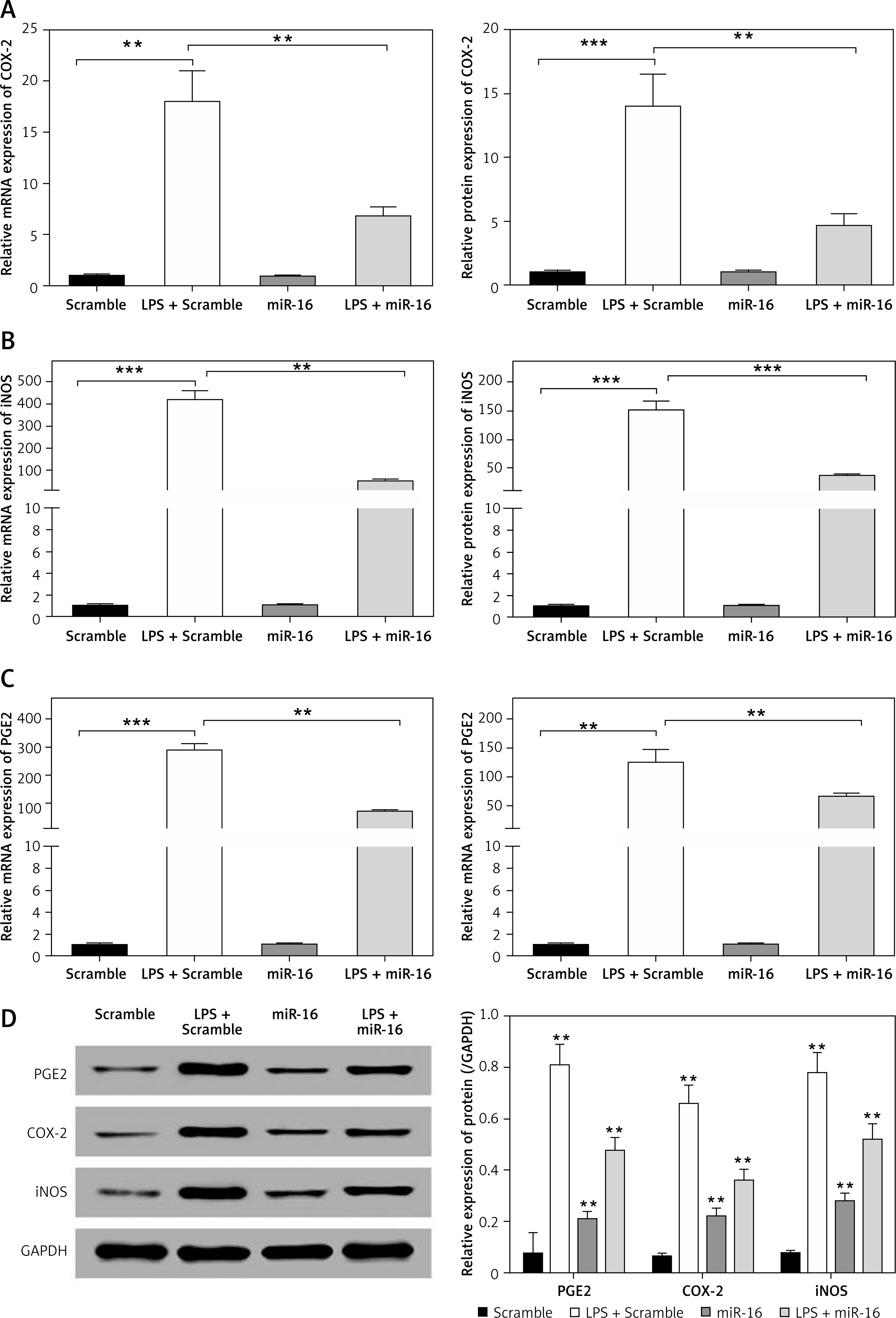
Overexpressed miR-16 inhibited the expression of nitric oxide-related genes
Nitric oxide (NO) and PGE2 are important pro-inflammatory mediators, which can lead to neuralgia and promote IDD progression by changing the microenvironment of IVD via an inflammatory response [15]. To confirm the regulatory role of miR-16 in the expression of NO-related genes in LPS-induced NP cells, the experiment of expression determination was conducted. The results suggested that the gene expression of COX-2, iNOS and PGE2 in the NP cells was significantly upregulated by LPS (p < 0.01, p < 0.001) and significantly downregulated by overexpressed miR-16 (Figures 2 A–C). Through the ELISA experiment, we found that overexpressed miR-16 could significantly suppress the protein expression of COX-2, iNOS and PGE2 in LPS-induced NP cells (p < 0.01, p < 0.001) (Figures 2 A–C). Finally, western blot was performed to assess the expression of COX-2, iNOS and PGE2 in LPS-induced NP cells and the result was similar to the ELISA experiment (Figure 2 D).
Figure 2
MiR-16 suppressed the LPS induced NO response associated gene expression. A – MiR-16 suppressed LPS induced COX-2 expression (p < 0.01, p < 0.001). B – MiR-16 suppressed LPS induced iNOS expression (p < 0.01, p < 0.001). C – MiR-16 suppressed LPS induced PGE2 expression (p < 0.01, p < 0.001). D – Western blot showed the expression of PGE2, iNOS and COX-2 in different treatment (p < 0.01)
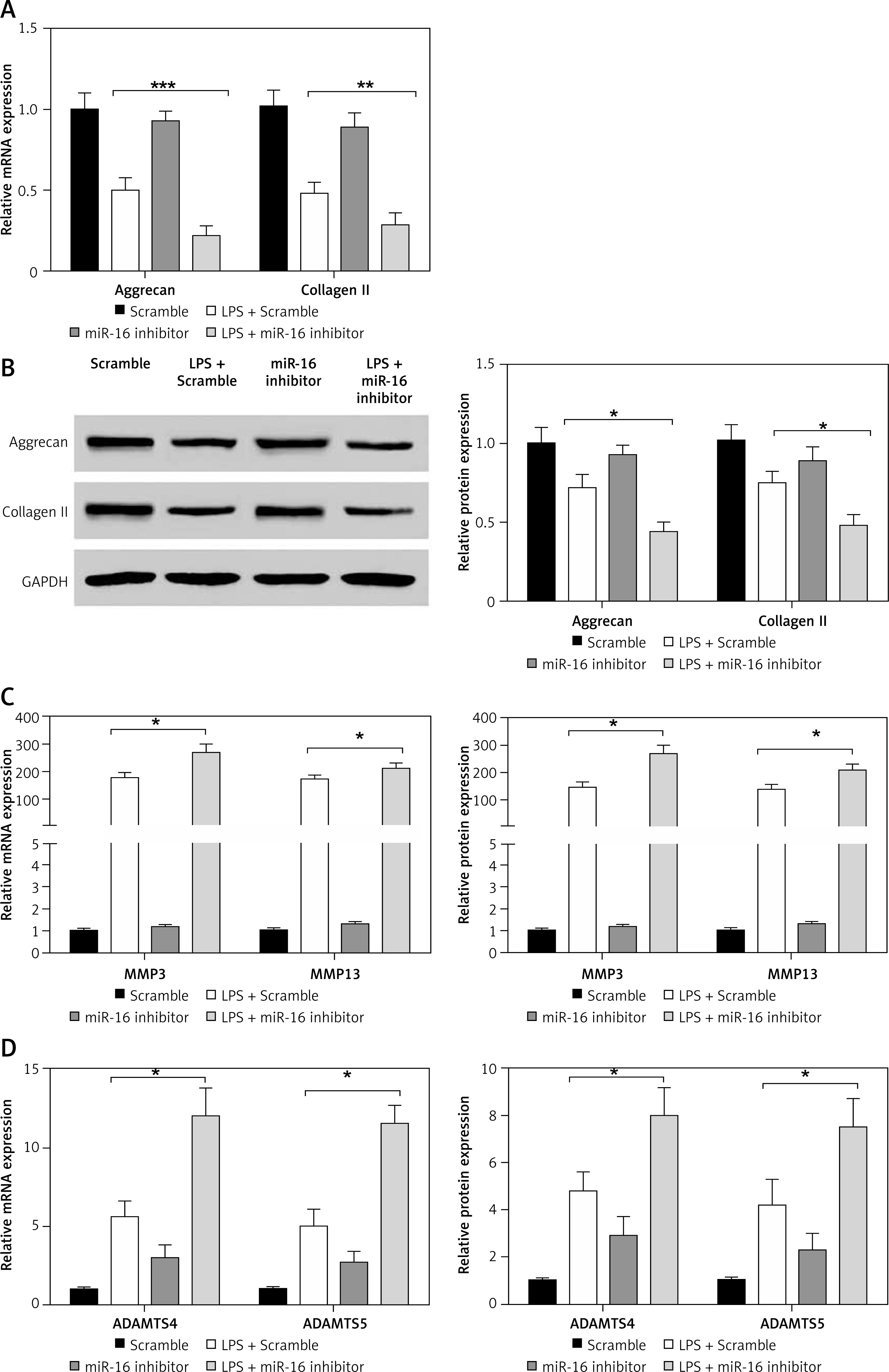
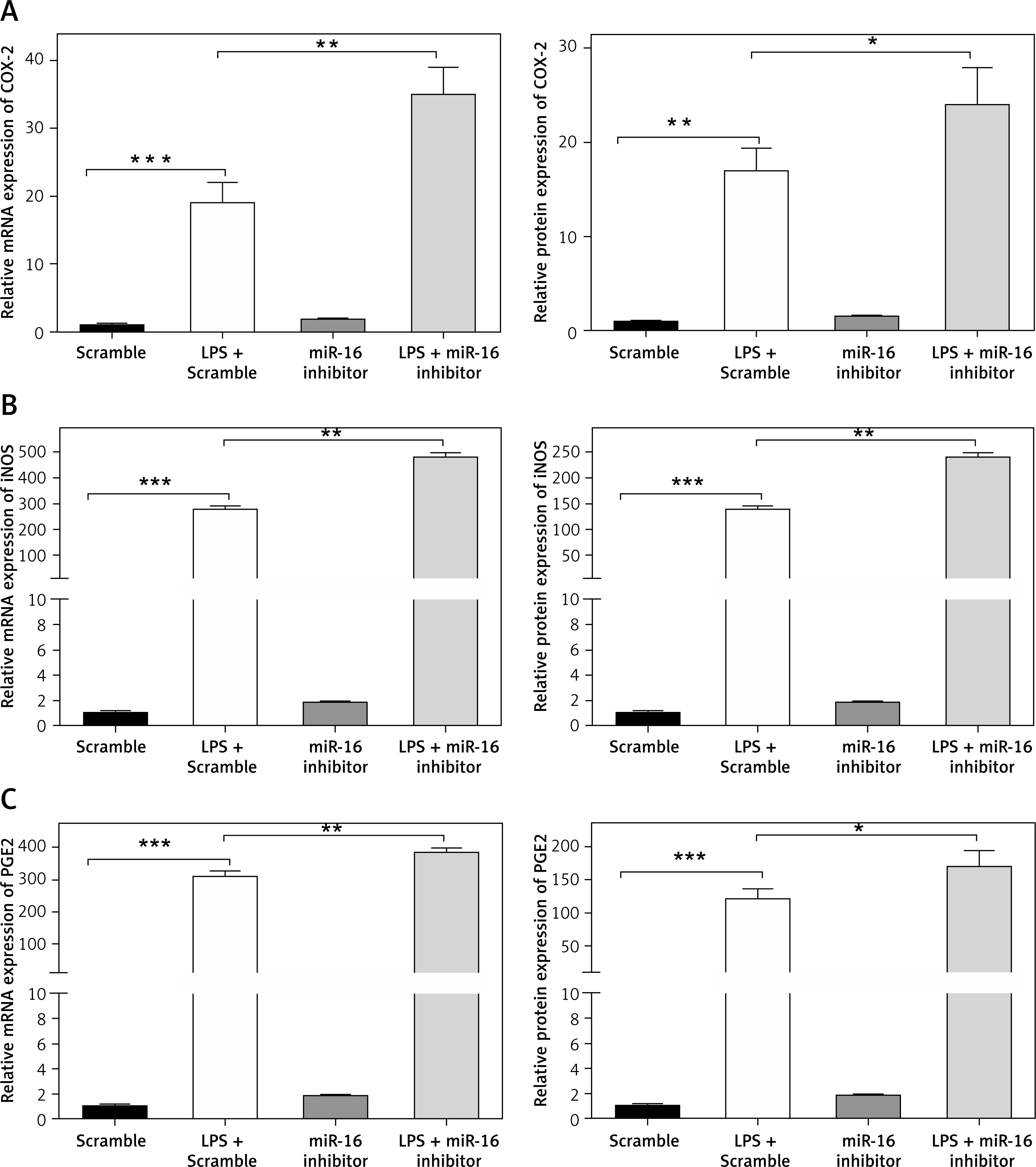
MiR-16 inhibitor regulated the expression of ECM genes and synthesis of ECM degrading enzymes
In order to further confirm the regulatory effect of miR-16 on the inflammatory response in LPS-induced NP cells, an miR-16 inhibitor was used to treat NP cells to block the normal function of miR-16. As shown in Figures 3 A and B, the miR-16 inhibitor intensified the inhibitory efficacy of LPS on the expression of aggrecan and collagen II in NP cells; western blotting results showed that the miR-16 inhibitor significantly decreased aggrecan and collagen II protein production after treatment with LPS (p < 0.05, p < 0.01, p < 0.001). By contrast, the miR-16 inhibitor significantly promoted the gene expression of ECM degrading enzymes, including MMP3, MMP13, ADAMTS4 and ADAMTS5, which were induced by LPS. In addition, the ELISA experiment further confirmed that the miR-16 inhibitor significantly promoted the protein expression of the above ECM degrading enzymes (p < 0.05) (Figures 3 C, D).
Figure 3
MiR-16 inhibitor suppressed the expression of extracellular matrix genes and stimulated the expression of ECM degradation enzymes. A and B – MiR-16 inhibitor decreased the mRNA and protein level of aggrecan and collagen II (p < 0.05, p < 0.01, p < 0.001). C – MiR-16 inhibitor promoted the mRNA and protein level of MMP3 and MMP13 (p < 0.05). D – MiR-16 inhibitor promoted the mRNA and protein level of ADAMTS4 and ADAMTS5 (p < 0.05)
MiR-16 inhibitor regulated the expression of NO-related genes
Monolayer cultures of NP cells were transfected with the synthetic miR-16 inhibitor and stimulated with LPS, followed by a PCR assay and ELISA to measure the mRNA and protein levels respectively of various matrix-degrading enzymes and proinflammatory factors. The MiR-16 inhibitor markedly promoted the mRNA levels of COX-2, iNOS and PGE2 induced by LPS, and the ELISA assay demonstrated that the miR-16 inhibitor increased the LPS-induced COX-2, iNOS and PGE2 production in NP cells (p < 0.05, p < 0.01, p < 0.001) (Figure 4).
Figure 4
MiR-16 inhibitor stimulated the expression of NO response associated genes. A – MiR-16 inhibitor promoted the mRNA and protein level of COX-2 (p < 0.05, p < 0.01, p < 0.001). B – MiR-16 inhibitor promoted the mRNA and protein level of iNOS (p < 0.01, p < 0.001). C – MiR-16 inhibitor promoted the mRNA and protein level of PGE2 (p < 0.05, p < 0.01, p < 0.001)
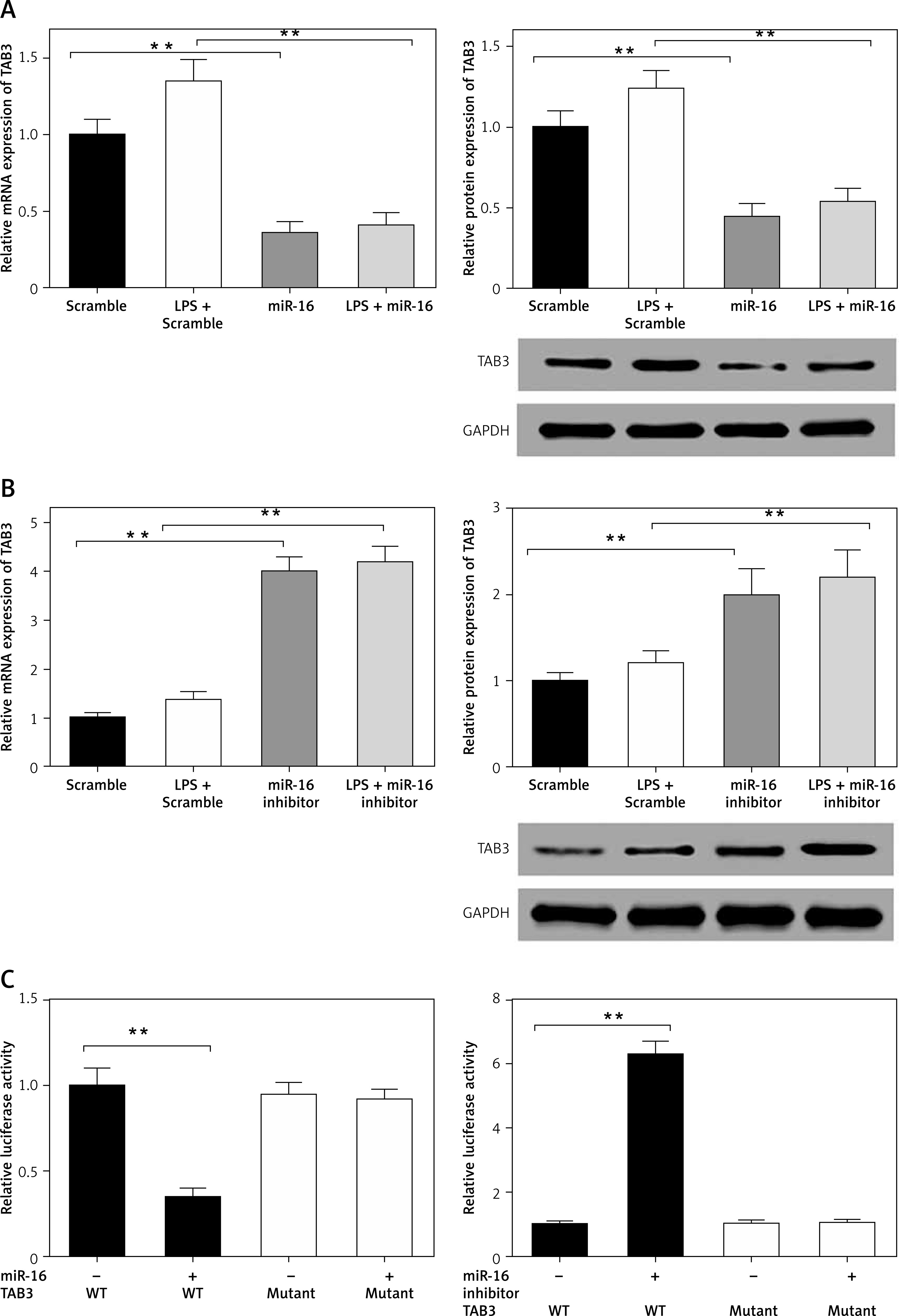
MiR-16 regulated TAB3 gene expression
Through target prediction, we found that miR-16 could target the TAB3 gene directly. It has been reported that TAB3 is relevant to cellular immunity and inflammatory responses. Therefore, we studied whether miR-16 could regulate gene expression of TAB3. The experiment results showed that overexpressed miR-16 led to significant downregulation of mRNA and the protein level of TAB3 in NP cells (p < 0.01, p < 0.001) (Figure 5 A). In contrast, the miR-16 inhibitor led to significant upregulation of mRNA and protein expression of TAB3 (p < 0.01, p < 0.001) (Figure 5 B). It indicated that miR-16 had an inhibitory effect on the gene expression of TAB3. We established the luciferase reporter plasmid of TAB3 3′-UTR sequence to further explain the direct effect of miR-16 on the TAB3 gene. The results showed that the TAB3 3′-UTR-mediated luciferase activity was significantly inhibited by miR-16, while it was significantly promoted by the miR-16 inhibitor (p < 0.01, p < 0.001); on the other hand, there was no significant difference in the mutant TAB3 3′-UTR-mediated luciferase activity (Figure 5 C).
Figure 5
MiR-16 regulated the gene expression of TAB3. A – MiR-16 suppressed mRNA and protein level of TAB3. B – MiR-16 inhibitor stimulated mRNA and protein level of TAB3. C – MiR-16 suppressed TAB3 5′-UTR derived luciferase activity but did not affect the luciferase activity of the mutant. MiR-16 inhibitor stimulated TAB3 5′-UTR derived luciferase activity, but did not affect the luciferase of the mutant
MiR-16 regulated NP cells through NF-κB and MAPK signaling pathways
The downstream signaling pathways of the TAK1-TAB2/3 complex include NF-κB and MAPK. Therefore, we hypothesized that the regulatory effect of miR-16 on NP cells is mediated via the NF-κB and MAPK pathways. Our study identified the regulatory effect of miR-16 on the NF-κB-mediated transcriptional activity through the transfection of the interference sequence. The results showed that miR-16 significantly inhibited the expression of p-p65; in contrast, the miR-16 inhibitor significantly promoted the expression of p-p65 (p < 0.01) (Figure 6). We measured the activation of the MAPK pathway related proteins by western blotting to evaluate the potential mechanisms of effects of miR-16 on LPS induced NP cells. The results showed that LPS significantly activated the MAPK pathways by promoting JNK, ERK and P38 protein phosphorylation. Also, the protein expression of p-ERK1/2, p-p38 MAPK and p-JNK was increased in the miR-16 inhibitor group, while it was decreased in the miR-16 mimics group; and there was no significant change in the total protein expression (Figure 6).

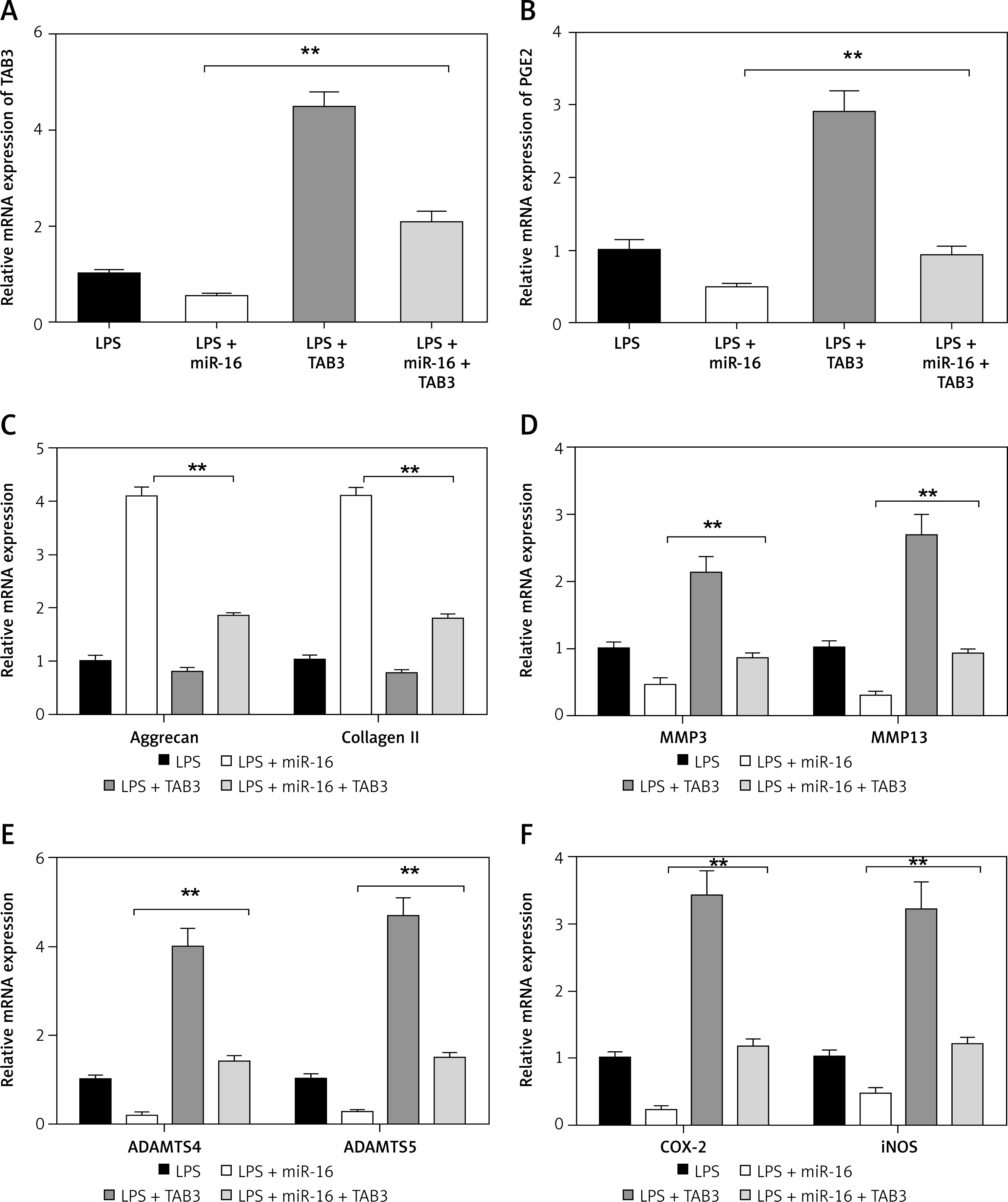
Overexpressed TAB3 reversed the regulatory function of miR-16 on NP cells
In terms of verifying whether miR-16 mediates the regulation of inflammatory responses in NP cells through the inhibition of TAB3, we detected the effect of overexpressed TAB3 on the regulatory function of miR-16. It was found that overexpressed TAB3 reversed the regulatory function of miR-16 on the expression of ECM genes and synthesis of ECM degrading enzymes, including blocking the promoting effect of miR-16 on aggrecan and collagen II expression (p < 0.01), blocking the inhibitory effect of miR-16 on the gene expression of MMP3, MMP13, ADAMPTS4, ADAMPTS5, and COX-2, iNOS, and PGE2 (p < 0.01) (Figure 7).
Figure 7
Overexpression of TAB3 reversed the regulatory function of miR-16 in LPS induced NP cells. A – Relative expression levels of TAB3 in LPS-induced NP cells transfected with miR-16, TAB3 or miR-194 + TAB3. B – Overexpression of TAB3 rescued the expression of PGE2 (p < 0.01). C – Overexpression of TAB3 reversed the miR-16 induced promotion of aggrecan and collagen II expression (p < 0.01). D – Overexpression of TAB3 rescued the expression of MMP3 and MMP13 which were promoted by miR-16 (p < 0.01). E – Overexpression of TAB3 rescued the expression of ADAMTS4 and ADAMTS5, which were suppressed by miR-16 (p < 0.01). F – Overexpression of TAB3 rescued the expression of COX-2 (p < 0.01) and iNOS (p < 0.01)
Discussion
IDD is a common chronic disease, frequently subjected to clinical study, which seriously threatens the quality of life of patients, while imposing an economic burden on society [16, 17]. Currently, relieving clinical symptoms is the main treatment method for IDD, instead of starting from the pathological mechanism, so there is still a lack of efficient treatment method of this disease. The function of microRNA in IDD has become a hot issue gradually in the last 5 years. The study indicated that microRNA is involved in the IDD process through multiple biological processes, mainly including regulating apoptosis [18] and proliferation [19] of NP cells, controlling composites [20] of ECM and inflammatory responses [21], and mediating chondral plate degeneration [22]. The multiple functions of the miRNA molecule in IDD provided a new idea and method for the treatment of IDD. Through exploring miR-16, this study further enriched knowledge of the functions of microRNA in IDD.
In this study, miR-16 was downregulated in LPS-induced NP cells, which indicated that miR-16 reacted to the inflammatory response. LPS inhibited the gene and protein expression of aggrecan and collagen, while miR-16 exerted the opposite effect. Further experiments proved that miR-16 could attenuate the inhibitory effect of LPS on aggrecan and collagen. Meanwhile, LPS promoted the gene and protein expression of ECM degrading enzymes (MMP3, MMP13, ADAMPTS4, ADAMPTS5), which was contrary to the function of miR-16. Additionally, overexpressed miR-16 could suppress PGE2 and NO expression induced by LPS, and the miR-16 inhibitor could up-regulate the expression of ECM genes and synthesis of ECM degrading enzymes. These findings revealed that miR-16 could inhibit the inflammatory response of NP cells induced by LPS. Therefore, miR-16 may be a novel mediator in the inflammatory response of NP cells, which will provide a new target for the therapeutic improvement of inflammatory responses in NP cells.
To explore the anti-inflammatory mechanism of miR-16 in NP cells, we applied two algorithm programs (TargetScan and miRBase) to search for potential targets of miR-16. It was found that TAB3 is a target of miR-16. In the NF-κB pathway, TAB3 can couple TAK1 and TRAF6 together as an adaptor protein. Subsequently, we found that TAB2 and TAB3 could marry with the ubiquitinated ‘lysine 63’ (K63) chains synthesized by TAK1 so as to activate the NF-κB pathway. Meanwhile, the TAK1-TAB2/3 complex can phosphorylate MAPKs (including MEKK3 and MEKK6), and MEKKs can in turn phosphorylate MAPKs to activate the transcription factor activator protein-1 (AP-1), which contributes to the expression of inflammatory cytokines [23, 24]. The present study showed that the TAB3 3′-UTR-mediated luciferase activity was significantly downregulated by overexpressed miR-16, while it was upregulated by the miR-16 inhibitor. Also, there was no significant difference in the mutant TAB3 3′-UTR-mediated luciferase activity. Additionally, overexpressed TAB3 plays an important role in reversing the effect of miR-16 on the expression of ECM genes and synthesis of ECM degrading enzymes. Thus, miR-16 exerts an inhibitory effect on the inflammatory response in LPS-induced NP cells by targeting TAB3 directly. Further, it was reported that LPS might be involved in coronary heart disease [25].
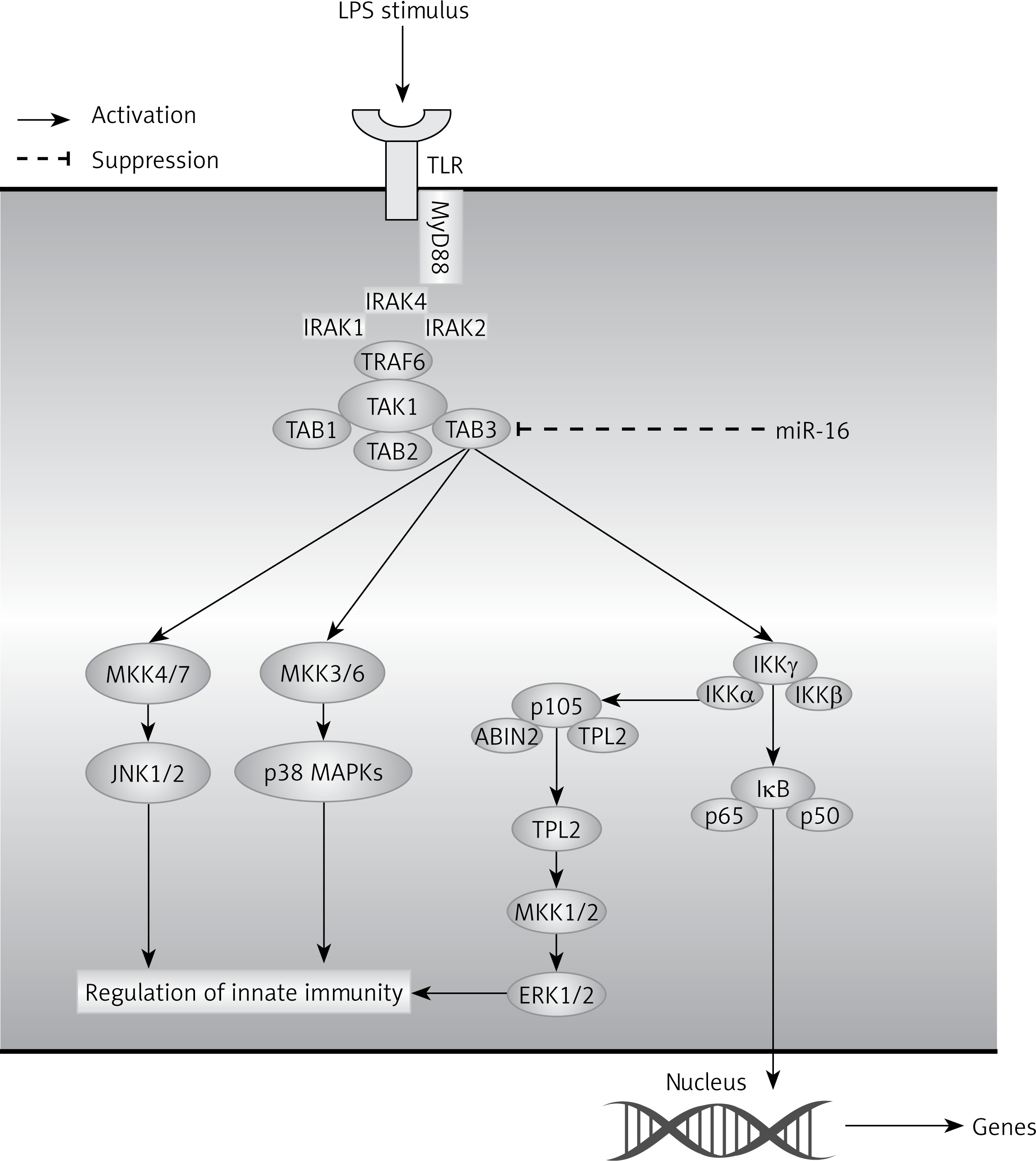
The downstream pathways of LPS mainly include NF-κB and MAPK. When cells are maintained under normal physiological conditions, the activity and functions of NF-κB in the cytosol will be inhibited; when cells are stimulated, NF-κB will be activated to transfer into the nucleus and mediate the expression of downstream genes, including inflammatory-associated factors (TNF-α, iNOS, and PEG, etc), and further regulate various inflammatory responses. In general, MAPK mainly includes three signaling pathways, JNK, ERK1/2 and p38 MAPK, which play substantial roles in the regulation of inflammatory mediators. When activated, the MAPK pathway can promote inflammatory responses by upregulating pro-inflammatory cytokines directly and by activating an NF-κB signal [26]. In addition, it has been demonstrated that activated p38 MAPK, ERK1/2 and JNK can also activate NF-κB [27, 28]. In a former study, we found that miR-16 regulated inflammatory responses of NP cells by targeting TAB3. Hence, our study investigated the association between miR-16 and NF-κB and the MAPK signaling pathway, which is downstream of TAB3. The results showed that overexpressed miR-16 inhibited the NF-κB luciferase activity, and that the miR-16 inhibitor promoted NF-κB luciferase activity. Additionally, the results of western blot showed that the protein expression of p-ERK1/2, p-p38 MAPK and p-JNK was upregulated by miR-16 inhibition, while it was downregulated by miR-16 mimics, which suggested that the regulatory function of miR-16 is mediated through NF-κB and MAPK pathways. In combination with previous experiments, we can learn that miR-16 might regulate inflammatory responses through NF-κB and MAPK signaling pathways (Figure 8).
Figure 8
A hypothetical model for the role of miR-16 in LPS induced inflammation in NP cells. After LPS stimulus in NP cells, TLR4 located in the cell membranes recruited MyD88 inside the cells and then recruited MyD88 bound together with IRAK. Following that, TRAF6 was activated by IRAK, then recruited and activated TAK1 via TAK1-associating chaperones, TAB2 and TAB3 (TAB2/3). TAK1 further phosphorylated the MAPK kinases and IKKβ. The MEKKs phosphorylated the MAPKs, leading to activation of the transcription factor AP-1, whereas IKKβ phosphorylated IκBα, leading to the release of NF-κB to the nucleus, which led to induction of various cytokines and chemokines and subsequent inflammatory responses. In addition, miR-16 could exert an inhibitory effect on NF-κB or MAPK signal pathways by targeting TAB3
In conclusion, miR-16 could inhibit the activation of NF-κB and MAPK signaling pathway by targeting TAB3, and further negatively regulate the LPS-induced inflammatory responses in NP cells. In this way, miR-16 is expected to be a potential target for the therapy of IDD.