Introduction
Gliomas are prevalent neuroepithelial tumours of the brain and are responsible for significant human mortality [1]. The gliomas are oligodendrocytic or astrocytic in origin or may sometimes be a mix of both these types. Gliomas account for approximately 70% of all brain tumours [2]. The mortality rate of gliomas is very high, and the survival rate is very poor. The patients with advanced-stage gliomas have an average survival of 16 months [3]. It has been reported that early diagnosis and development of efficient and novel chemotherapeutic agents may help to curb the incidence of gliomas and to improve the 5-year survival [4]. The utilisation of microRNAs (miRs) for cancer therapy is currently one of the promising fields in cancer research [5]. The miRs are endogenous molecules around 18–25 nucleotides long that control the expression of the target genes by binding to 3´UTR causing degradation of mRNA or repression of translation [6]. The miRs play vital roles in fundamental cellular processes, which include but are not limited to proliferation, development, differentiation, apoptosis, and autophagy [7]. There is strong evidence that many miRs show dysregulation in cancerous tissues and play an important part in the development of cancer [8]. Hence, it is believed that miRs may serve as therapeutic targets and will allow targeted therapy for the treatment of cancer [9]. MiR-16 has been shown to control the growth of different cancers types. The regulation of bladder cancer growth by miR-16 via modulation of cyclin B1 is reported in the literature [10]. MiR-16 suppresses the proliferation of colorectal cancer cells via targeting of p53/survivin [11]. In another study, miR-16 promoted apoptosis in cancer cells by suppressing Bcl-2 [12]. MiR-16 has also been shown to regulate the drug resistance in gastric cancer cells [13]. However, the role of miR-16 via modulation of PI3K/AKT/mTOR signalling pathway has not been studied in gliomas. This study investigated the function and therapeutic utility of miR-16 in gliomas. Taken together, we miR-16 acts a tumour suppressor in glioma may point to a novel therapeutic target in glioma.
Material and methods
Glioma tissues, cell lines, and culture conditions
Glioma tissue specimens and normal adjacent tissues of 15 breast cancer patients who underwent surgical resection were obtained after informed consent in the Department of Neurosurgery, Jining No.1 People’s Hospital, Shandong, China from June 2018 to January 2019 were included. Patients were aged 31–67 years with an average age of 39.5 years. The study was approved by Ethics Committee of the institute under approval number JPH/66/2019. The glioma cell lines (U87, U118 MG, M059K, Hs683) and normal astrocytes were procured from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. The cells were cultured in RPMI-1640 medium containing 10% foetal bovine serum, 100 μg/ml streptomycin, 100 U/ml penicillin, and a humidified atmosphere containing 5% CO2.
The cDNA synthesis and qRT-PCR
The TRIzol reagent (Invitrogen/Life Technologies, Carlsbad, CA, USA) was used for the extraction of RNA from the tissues and cell lines. Subsequently, the total RNA was reverse transcribed by RevertAid cDNA synthesis kit (Fermentas). Thereafter, the quantitative RT-PCR was performed by using SYBR Green master mix (Applied Biosystems; Foster City, Calif, USA) on an ABI 7900HT system. The reaction mixture consisted of 20 μl containing 1.5 mM MgCl2, 2.5 U Taq DNA Polymerase, 200 μM dNTP, 0.2 μM of each primer, and 0.5 μg DNA. The cycling conditions were as follows: 95°C for 20 s, followed by 40 cycles of 95°C for 15 s, and 58°C for 1 min. GAPDH was used as an internal control, and the relative quantification (2–ΔΔCq) method was used to evaluate the quantitative variation between the samples. All reactions were carried out in triplicate, and the gene expression was determined by the 2−ΔΔCt method.
Cell transfection
The miR-16 mimics and miR-NC were ordered from RiboBio (Guangzhou, China). Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) was then used to perform the transfections in accordance with the manufacturer’s guidelines. As the U118 MG cells reached 80%, the appropriate concentrations of miR-151 mimics or miR-NC was transfected into these cells.
Cell proliferation assay
The miR-NC and miR-16 mimic-transfected U87 MG cells were seeded in white bottom 384-well plates (Thermo-scientific) at a density of 5 × 102 cells/well in 25 μl medium and subjected to incubation at 37°C for 24 h. Thereafter, live cell counts were measured by CellTiter-Glo assay (Promega, Madison, WI) in accordance with user guidelines. An Envision Multi-Label Reader (Perkin Elmer, Waltham, MA) was employed to measure the luminescence produced by the live cells at different time intervals (0, 12, 24, 48, and 96 h). For each time point, the U118 MG cell percentage was plotted. The experiments were performed in triplicate.
Colony formation assay
The U118 MG cells (2.5 × 105/well) were transfected with miR-NC and miR-16 mimics and incubated for 24 h at 37°C. Thereafter, the cells were trypsinised and subjected to washing with phosphate-buffered saline (PBS). The U118 MG cells with 500 cells/well were seeded in 12-well plates and subjected to culturing for 10 days. This was followed by fixation of the cells with 4% paraformaldehyde and stained with 0.1% crystal violet. The cell colony images were obtained by using an inverted microscope.
Apoptosis assays
The U118 MG cells were transfected with suitable constructs and cultured for 24 h at 37°C and then fixed with ethanol (70%) for 20 min. The cells were then subjected to PBS washing and subsequently stained with a DAPI or a solution of AO and EB. Finally, the cells were examined under microscope to detect the induction of apoptosis. The U118 MG cells were transfected with appropriate constructs and then incubated for 48 h at 37°C. The cells were subsequently dissociated with the help of trypsin and then PBS washed. The cells were then resuspended in 1X binding buffer, which was followed by the addition of 5 μl of annexin V-FITC and propidium iodide (PI). The cell culture was then placed in dark room for 15 min, after which the apoptosis percentage was evaluated by a flow cytometer.
Wound healing assay
The transfected U118 MG cells were placed in 12-well plates with approximately 1 × 105 cells per well. A scratch was made by a pipette tip at 24 h after transfection. The cells were PBS washed and fresh media was added. Following 24 h incubation at 37°C, the cells were subjected to fixation with methanol. The initial and final wound width was determined from photomicrographs.
Cell invasion assay
Transwell chambers with Matrigel were used to monitor the U118 MG cell invasion. Briefly, the cells were transfected with appropriate constructs, and 48 h post-transfection the cells were harvested and suspended in fresh media. While 200 μl of the cell suspension containing approximately 5 × 104 cells was placed in the upper compartment, 500 μl of fresh media was placed in the lower compartment. After 24 h, cells present at the upper compartment were removed by swabbing, while cells that invaded the lower surface were fixed and then subsequently stained with 0.05% crystal violet. Finally, 10 random fields were selected to determine the invasion under a light microscope.
Western blotting
The glioma tissues and cell lines were lysed, and the protein concentration in each sample was measured by Bradford assy. Equal concentrations of the proteins from each sample were loaded on 10% SDS polyacrylamide gel and then followed by shifting to polyvinylidene fluoride membranes. Blocking of the membrane was then performed using fat-free milk (5%) in TBST. This was followed by incubation with a primary antibody for 24 h at 4°C. Subsequently, a secondary antibody was added at 25°C for about 2 h. The bands of interest were finally observed by chemiluminescence.
Statistical analysis
The experiments were carried out in three biological replicates. The data are presented as the mean of the replicates ± SD. The statistical analysis utilised Student’s t-test or one-way ANOVA followed by Tukey’s test. P < 0.05 was considered as a statistically significant difference.
Results
MiR-16 is suppressed in glioma cells
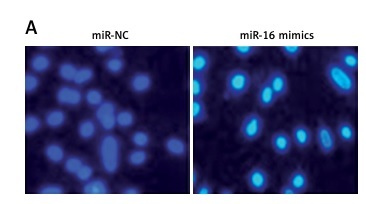
The miR-16 expression was determined in the glioma tissues and in the normal adjacent tissues of 15 patients. The qRT-PCR results revealed that miR-16 expression was significantly suppressed in glioma tissues in comparison to the adjacent normal tissues (Figure 1 A). Gene expression analysis of miR-16 in glioma cells showed miR-16 to be significantly (up to eight-fold) downregulated in glioma cell lines (Figure 1 B). The expression of miR-16 was highly and significantly suppressed in U118 MG cells. This cell line was therefore used for further studies.
Figure 1
miR-16 inhibits the proliferation of cells. A – Expression of miR-16 in glioma and normal adjacent tissues. B – Expression of miR-16 in different glioma cell lines. C – Expression of miR-16 in miR-NC or miR-16 mimic-transfected U118 MG glioma cells. D – Cell survival (%) of miR-NC or miR-16 mimic-transfected glioma U118 MG cells. E – Colony formation of the miR-NC or miR-16 mimic-transfected glioma U118 MG cells. The experiments were performed in triplicate and expressed as mean ± SD (*p < 0.05)
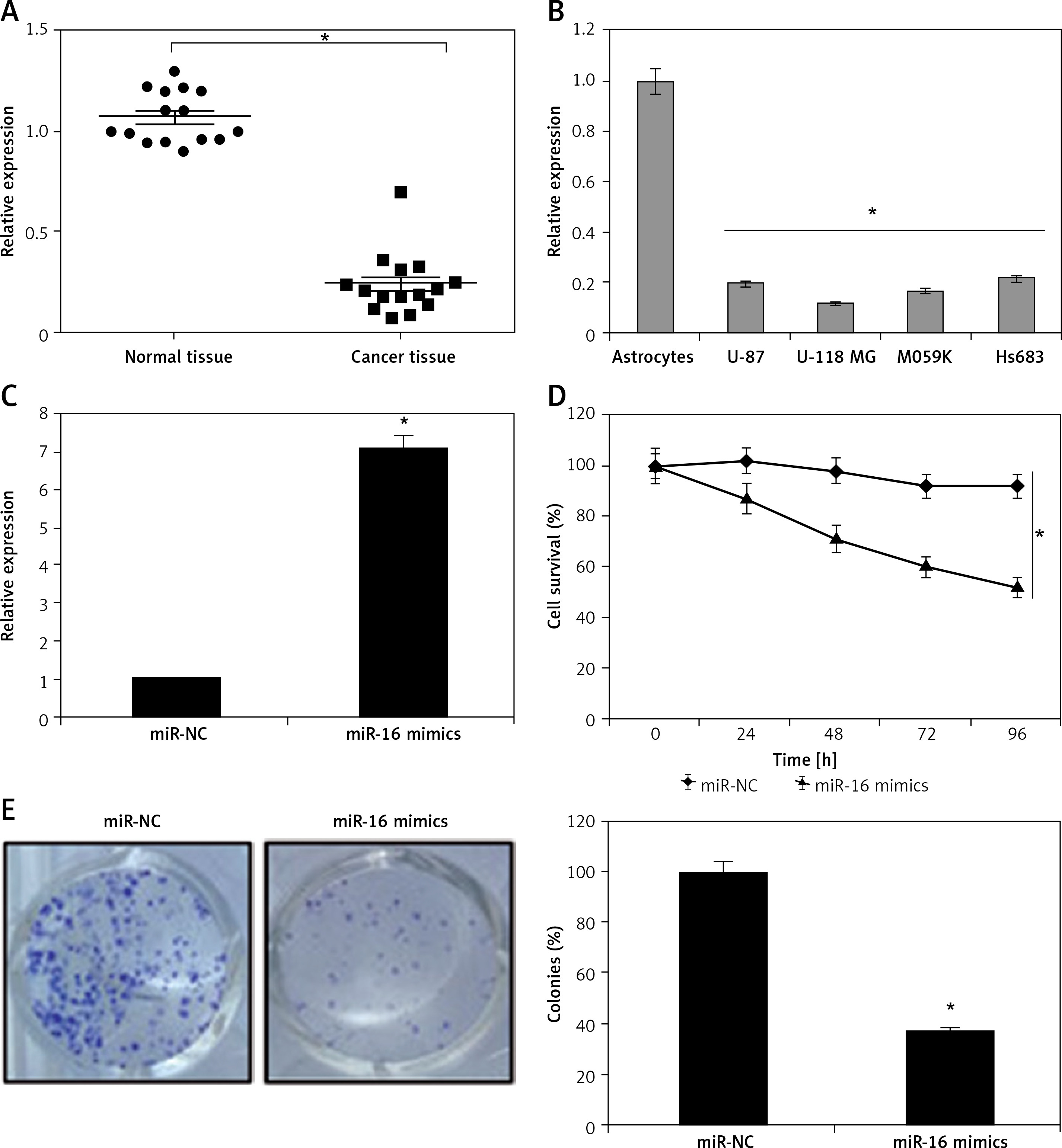
MiR-16 induces apoptosis in U118 MG glioma cells
The miR-NC and miR-16 mimic-transfected cells were subjected to cell count assay. The findings showed that miR-16 overexpression suppressed the proliferation of the U118 MG cells. To decipher the underlying molecular mechanism for the miR-16 induced inhibition of cell growth, we performed DAPI and AO/EB staining of cells. The DAPI staining assay showed that miR-16 overexpression increased the DAPI-positive cells. The AO/EB staining showed that miR-16 overexpression promoted the nuclear fragmentation of the U118 MG cells. The annexin V/PI staining showed that miR-16 overexpression increased the apoptotic U118MG cells from 1.86% in miR-NC to about 2.02% in miR-16 mimics. The miR-16 also enhanced the expression of Bax and depleted the expression of Bcl-2, further confirming the apoptotic cell death (Figure 2).
Figure 2
MiR-16 induces apoptosis in U118 MG cells: DAPI (A), AO/EB (B), and annexin V/PI staining (C) of miR-NC and miR-16 mimic-transfected U118 MG transfected cells. D – Western blots showing the expression of Bax and Bcl-2 in miR-NC and miR-16 mimic transfected U118 MG transfected cells. The experiments were performed in triplicate and expressed as mean ± SD (*p < 0.05)
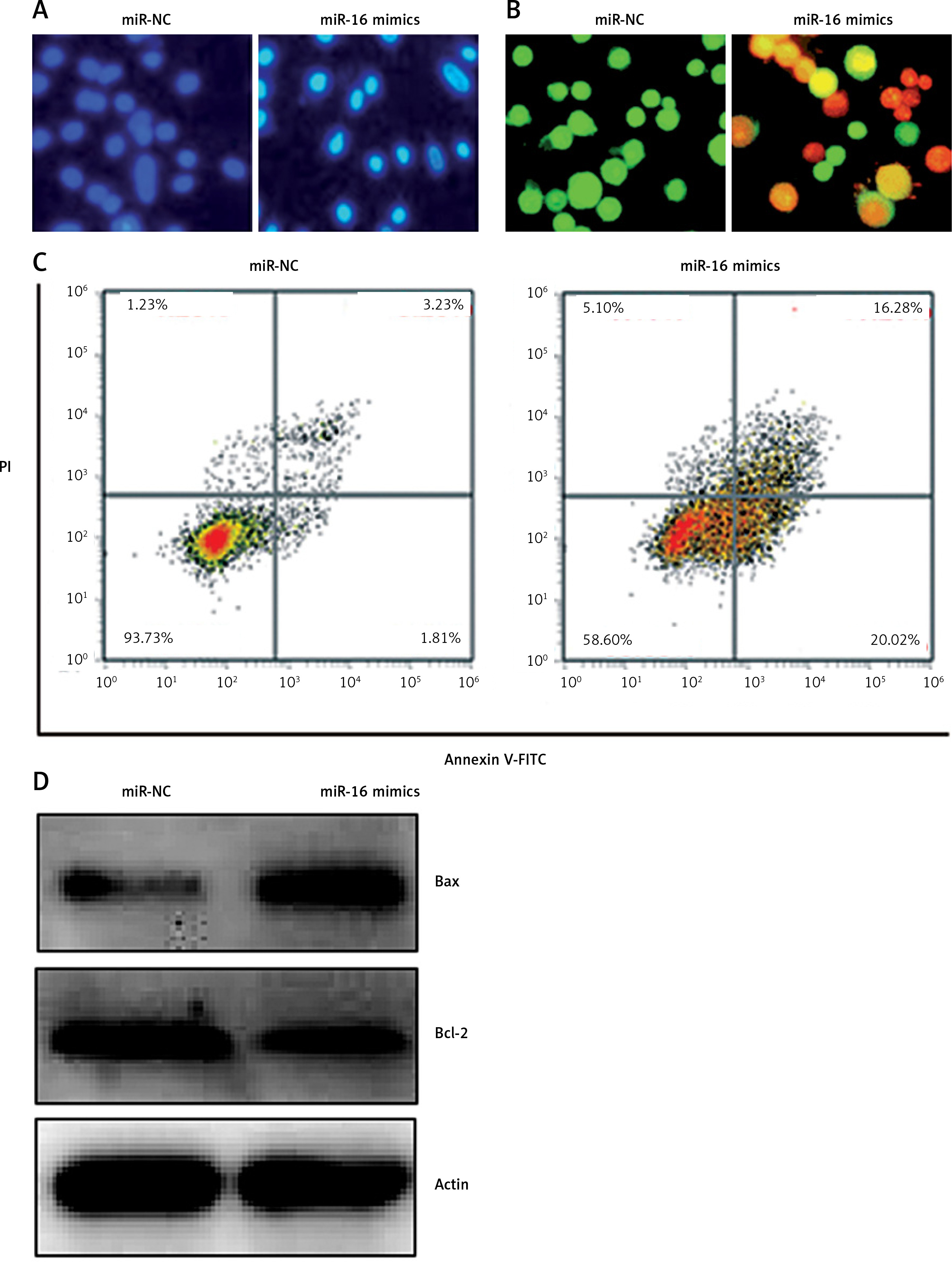
MiR-16 suppress the metastasis of U118 MG cells
The effects of miR-16 on the migration of the U118 MG cells were determined by wound healing assay. The results showed that miR-16 caused a significant decrease in the migration of the cells, as evident from the wound closure (Figure 3). Transwell assay was used to investigate the cell invasion of the U118 MG cells, and the results showed that the migration of the U118 MG cells was significantly inhibited. The invasion in the case of miR-16 overexpressing U118 MG cells was found to be 40% relative to control (Figure 4 A). The inhibition of cell invasion also accompanied with the downregulation of MMP-2 and MMP-9 expression in miR-16 mimic-transfected U118 MG cells (Figure 4 B).
Figure 3
Wound healing assay showing migration of the miR-NC and miR-16 mimic-transfected U118 MG cells. The experiments were performed in triplicate and expressed as mean ± SD (*p < 0.05)

Figure 4
A – Transwell assay showing the invasion in of the miR-NC and miR-16 mimic-transfected U118 MG cells. B – Western blot analysis showing expression of MMP-2 and MMP-9 in miR-NC and miR-16 mimic-transfected U118 MG cells. The experiments were performed in triplicate and expressed as mean ± SD (*p < 0.05)
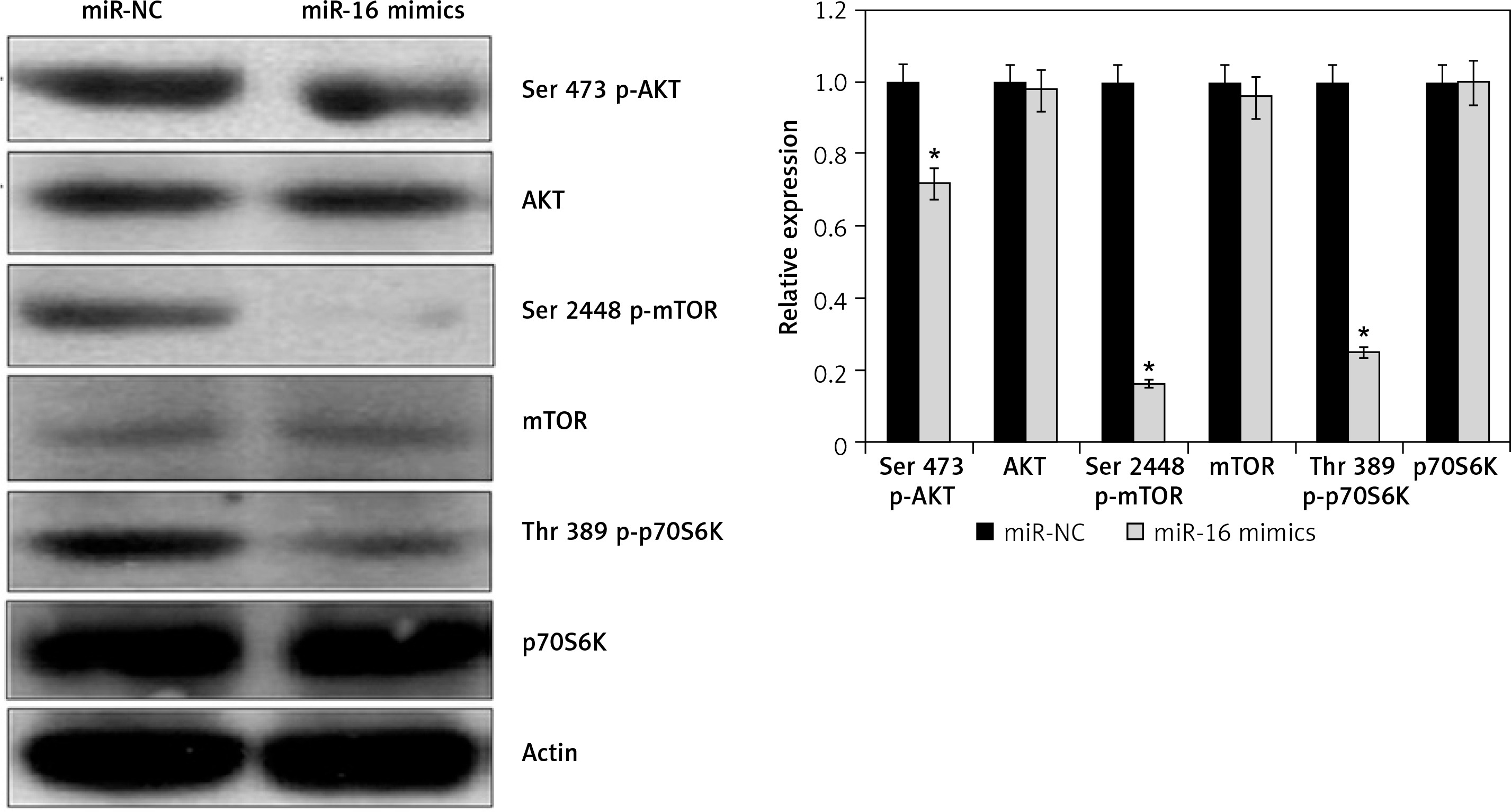
Effect of miR-16 on PI3K/AKT/mTOR signalling pathway
The effects of miR-16 overexpression were examined on the PI3K/AKT in the U118 MG cells. The results showed that miR-16 overexpression inhibited the phosphorylation of the p70S6K, AKT, and mTOR at Ser473, Ser2448, and Thr389, respectively, in U118 MG cells (Figure 5). Nonetheless, total p70S6K, AKT, and PI3K levels were found to be unchanged upon miR-16 overexpression.
Discussion
Gliomas are prevalent brain tumours in adults occurring in any part of the central nervous system [14]. Although gliomas are generally malignant, some of them may not always behave as malignant [15]. Being aggressive types of brain tumours, approximately 18,000 new glioma cases and 13,000 deaths are reported in the USA annually [16]. The limited availability of reliable and efficient therapeutic targets/agents hinders the treatment of gliomas [17]. MiRs have shown great promise as therapeutic agents for cancer treatment [18]. Since the discovery of first miR in the early 1990s, several miRs have been reported to play vital roles in different cellular processes, such as proliferation, cell death, and metastasis via modulation of post-gene expression post-transcriptionally [19]. MiRs have been shown to act as tumour suppressors as well as oncomiRs, and many miR-based therapies are being evaluated for cancer treatment [20]. MiR-16 has been found to act as a tumour suppressor, but its therapeutic implications via regulation of PI3K/AKT/mTOR cascade have not been explored in gliomas. Herein, we determined the therapeutic implications of miR-16 in gliomas and also examined its effects on the PI3K/AKT/mTOR pathway. We found that miR-16 expression was considerably suppressed in gliomas. These findings are complimented by earlier studies wherein miR-16 was found to be downregulated in colorectal cancer [21]. Apoptosis is an important mechanism by which defective and cancerous cells are removed from the body of the organism. Overexpression of miR-16 revealed that miR-16 inhibits the proliferation of U118 MG cells by induction of apoptosis. Earlier studies reported that miR-16 overexpression inhibited the proliferation of human breast cancer cells via induction of apoptosis [22]. The induction of apoptosis is also associated with alteration in the expression of several biomarker proteins [23]. Herein, we found that miR-16 overexpression caused an upsurge of Bax and downregulation of Bcl-2, further confirming the induction of apoptosis. The wound healing and transwell assays showed that miR-16 overexpression suppressed the metastasis of the glioma cells. These findings are in agreement with a previous study wherein miR-16 was reported to inhibit the migration and invasion of laryngeal carcinoma cancer cells [24]. Previous studies have shown that miR-16 overexpression inhibits the growth of hepatocellular cancer cells via modulation of the PI3K/AKT/mTOR signalling pathway [25]. Phosphatidylinositol 3-kinase (PI3K), Akt, and the mammalian target of rapamycin (mTOR) (PI3K/Akt/mTOR) signalling pathway are among the prevalently dysregulated cascades in a number of cancer types [26]. mTOR is a serine/threonine kinase, downstream of Akt, which regulates the growth and survival [27]. mTOR has been found to be a subunit of two distinct multi-protein complexes: mTORC1 and mTORC2. mTORC1 causes the phosphorylation of S6 ribosomal protein kinase (p70S6K) as well as initiation factor 4E-binding protein 1 (4EBP1) [28]. The 4EBP1 plays an essential role in the regulation of translation. mTORC2 is responsible for the phosphorylation of AKT at Ser473 [29]. The dysregulated activation of PI3K/AKT is responsible for poor response to therapeutic treatments and also enhances the drug resistance of several cancer types [30]. Therefore, the effects of miR-16 overexpression were examined on the PI3K/AKT/mTOR signalling pathway. The results showed that miR-16 overexpression inhibits the phosphorylation of p70S6K, AKT, and mTOR, suggesting the potential of miR-16 as therapeutic target for the management of gliomas.
In conclusion, the results suggest that that miR-16 is suppressed in glioma and its overexpression inhibits the growth and metastasis of glioma cells via modulation of PI3K and AKT signalling pathway. Taken together, miR-16 may be utilised as a therapeutic target for glioma treatment.