Introduction
Lung cancer, predominantly non-small-cell lung cancer (NSCLC), is the chief culprit for cancer mortality [1]. It is characterized by progressive metastasis and poor prognosis [2]. It is reported that lung cancer accounts for more than one-quarter (26%) of all cancer deaths, and the 5-year survival of lung cancer is only 18% [3]. Over the past decades, targeted therapeutics have been developed against drivers of lung adenocarcinoma, a subtype of NSCLC, improving the prognosis of patients [4]. Therefore, it is essential to further explore the underlying mechanisms of lung adenocarcinoma development and develop more efficient targeted treatment.
MiRNAs widely involved in cellular processes via bind to the 3′-untranslated region (3′-UTR) of genes and regulate cell functions [5, 6]. MiR-338-3p, located on chromosome 17q25, is a miRNA which plays a critical role in promoting cell death, neuronal differentiation and neurite extension [7]. Accumulating evidence has indicated that miR-338-3p could suppress the development of various human cancers, including hepatocellular carcinoma [8], breast cancer [9] and lung cancer [10]. It plays an anti-tumor role in lung cancer, inhibiting proliferation, migration, invasion, enhancing cell apoptosis [11] and suppressing tumor metastasis [12, 13]. Several studies have reported that several oncogenes were downregulated by miR-338-3p in lung cancer, such as IRS2 [10], SphK2 [14] and SOX4 [15]. Further investigation was needed to find more related genes for better understanding of lung cancer development.
AKAP12 is an A-kinase scaffold protein that possesses a characteristic binding domain for the regulatory subunit of protein kinase A [16, 17]. The loss of AKAP12 is associated with increased cancer susceptibility [16]. AKAP12 functions differently in various types of cancers. Xia et al. found that AKAP12 was inhibited by miR-103 in hepatocellular carcinoma, thus promoting tumor growth [18]. However, other studies suggested that overexpression of AKAP12 predicted a poor prognosis and promoted the tumor development in ovarian cancer [19] and melanoma [20]. Although evidence has highlighted that expression of AKAP12 was closely related to cancer development, current studies on its biological function in lung cancer are limited, and the interaction between miR-338-3p and AKAP12 also has not been determined.
In the present study, we investigated whether AKAP12 influenced the proliferation, migration, invasion and apoptosis in lung adenocarcinoma, and examined the relationship between AKAP12 and miR-338-3p. Our results suggested that miR-338-3p improved the prognosis of lung adenocarcinoma by suppressing AKAP12, providing a potential target for clinical treatment of lung adenocarcinoma.
Material and methods
Human specimens
In this study, 20 paired human lung adenocarcinoma tissues and adjacent non-tumor ones were obtained from the Affiliated Hospital of Hebei University of Engineering Hospital (Handan, China). No tissues were gained from patients who had received preoperative radiotherapy or chemotherapy before. Specimens were immediately frozen in liquid nitrogen and stored at –80°C until usage. The ethics committee of the Affiliated Hospital of Hebei University of Engineering approved this study.
Cell line and cell culture
Lung adenocarcinoma cell lines A549 and H1299 and normal human epithelial cell line HBE were purchased from BeNa Culture Collection (Beijing, China). Lung adenocarcinoma cell line A549 and normal cell line HBE were cultured in 90% Dulbecco’s modified eagle medium (DMEM, Sigma, St. Louis, MO, USA) with high glucose supplemented with 10% FBS, while H1299 was maintained in Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma) with 10% FBS. Cells were all cultured in 5% CO2 at 37°C.
Bioinformatics analysis
Microarray analysis was performed with the TCGA database, and 58 adjacent tissues and 58 tumor tissues in lung adenocarcinoma were involved respectively. Differentially expressing genes were recognized by an adjusted p-value less than 0.05 and log2 fold change larger than 10 or less than –10. After being ranked, the ten most highly expressed genes and most lowly expressed genes were listed in Supplementary Table SI. The heat map and volcano plot were drawn with R studio.
TargetScan and miRanda were used to predict the target relationship between AKAP12 and different miRNAs. Variable scores in the above two websites were recorded as shown in Supplementary Table SII: the mirSVR score in miRanda is an estimate of the miRNA effect on the mRNA expression level. The more negative the score, the greater the effect; the PhastCons score measures the conservation of nucleotide positions and the higher the score, the stronger the conservation; the Context++ score percentile indicated the percentage of sites for the miRNA with a less favorable context++ score, which is the sum of the contribution of 14 features [21], and a higher percentile means a greater combination probability.
RNA Folding Form website was used to predict the secondary structure of mRNAs. A greater structure change indicated a greater combination probability.
Immunohistochemistry
Lung adenocarcinoma tissue sections were firstly deparaffinized and rehydrated by ethanol solutions. After being washed by PBS 2-3 times, they were incubated with 3% H2O2 for 10 min and then were washed again with PBS. Sections were then immersed in 0.01 M citrate buffer (pH 6.0), heated by microwave at 95°C for 20 min, and cooled down at room temperature before cool water washing. They were then incubated with 3% goat serum sealant after PBS washing at room temperature for 20 min. Sections were firstly incubated overnight with primary rabbit anti-human antibody, anti-AKAP12 (1 : 200, ab198895, Abcam, Cambridge, MA, USA) at 4°C. Then they were washed in PBS and incubated with secondary antibody, goat anti-rabbit IgG-HRP (1 : 1000, ab6721, Abcam) at room temperature for 30 min. Following the final PBS washing, they were exposed to newly prepared diaminobenzidine for 4–6 min for 15 s of hematoxylin staining, in advance of dehydration by ethanol and air drying for observation. According to the positive staining intensity in immunohistochemistry assay, we set it to be that: colorless is score 0 (–), pale yellow is score 1 (+), palm yellow and above is score 2 (++). Six fields were randomly chosen.
Cell transfection
NC mimics, miR-107 mimics, miR-613 mimics, miR-206 mimics, miR-338-3p mimics, AKAP12-pcDNA3.1, sh-AKAP12-pcDNA3.1 and pcDNA3.1 plasmid vector were all obtained from GenePharma (Shanghai, China). Before transfection, cells were incubated in a 6-well plate in 5% CO2 at 37°C for 18–24 h. Lung adenocarcinoma cells were firstly transfected with pcDNA3.1 plasmid vector as the control, AKAP12-pcDNA3.1 as the AKAP12 group, and sh-AKAP12-pcDNA3.1 as the sh-AKAP12 group, respectively. Cancer cells were also transfected with 100 nM NC mimics, miR-107 mimics, miR-613 mimics, miR-206 mimics or miR-338-3p mimics respectively by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Lung adenocarcinoma cells comprised four groups: cells co-transfected with 50 nM NC mimics and 2 μg of pcDNA3.1 plasmid vector were the control group, cells co-transfected with 50 nM NC mimics and 2 μg of AKAP12-pcDNA3.1 were the AKAP12 group, cells co-transfected with 50 nM miR-338-3p mimics and 2 μg pcDNA3.1 plasmid vector were the miR-338-3p mimic group, and cells co-transfected with 2 μg of AKAP12-pcDNA3.1 and 50 nM miR-338-3p mimics were the AKAP12 + miR-338-3p mimic group.
Quantitative reverse transcription PCR
Total RNA was extracted from cells by TRIzol reagent (Takara Bio, Inc., Otsu, Japan). Reverse transcription of extracted RNA and real-time PCR were performed using the Reverse Transcription Kit (Takara Bio) and miRNAs qPCR Quantitation Kit (Takara Bio) according to the instructions of the manufacturer, with GAPDH as an internal control. Data were analyzed by StepOne Software (Applied Biosystems, Foster City, CA, USA). The relative expression level was expressed by 2–ΔΔCt . Primers for quantitative reverse transcription PCR (qRT-PCR) were synthesized by Huada Gene (Shenzhen, China) and listed in Table I.
Table I
Sequences for RT-qPCR, PCR, siRNA and mimics
Western blot
Total protein was extracted using ice-cold lysis buffer (Beyotime, Shanghai, China). Proteins were separated by SDS-PAGE with an electrophoresis system (Bio-Rad, Hercules, CA, USA), and then were transferred to polyvinylidene fluoride (PVDF) membrane (Invitrogen). After being blocked with 5% skimmed milk for 4 h, the PVDF membrane was incubated with primary rabbit anti-human antibodies, anti-AKAP12 (ab198895, 1 : 1000, Cambridge, MA, USA) and anti-GAPDH (ab9485, 1 : 2500, Cambridge, MA, USA). After incubation overnight at 4°C, the membrane was then incubated with secondary antibody, goat anti-rabbit IgG-HRP (ab6721, 1 : 2000), for 1.5 h. Protein expression was then visualized by ECL Plus Western Blotting Substrate (Thermo Scientific, Waltham, MA, USA). The concentration of proteins was quantified by Quantity One Software (Bio-Rad).
Clone formation assay
Cells were seeded in plates (200–300 cells per plate) and incubated in 5% CO2 at 37°C for 1–2 weeks until cell colonies were visible. After PBS washing, they were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for observation. Finally, clone number was quantified with a microscope.
MTT assay
Approximately 2 × 103 cells were seeded into 96-well plates for 3–5 days of incubation after suspension. Then 10 μl of MTT (5 mg/ml, Sigma) was added into each well for another 4 h of incubation. Supernatants were removed and 100 μl of dimethyl sulfoxide (DMSO, Sigma) was added to each well for 10 min shaking. Then the absorbance (optical density) was measured at 490 nm on a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Transwell assay
The upper chamber of a transwell chamber (Costar, Corning Inc., Corning, NY, USA) was plated with approximately 1 × 105 cells without serum, while the lower chamber was plated with 800 μl of culture medium (5 mg/l fibronectin and 10% FBS). After incubation at 37°C in 5% CO2 for 24 h, non-invading cells were removed from the upper chamber. Three independent fields were selected to observe hematoxylin and eosin stained bottom cells.
Flow cytometry (FCM) analysis
Transfected lung adenocarcinoma cells A549 and H1299 were incubated for 48 h, and then stained with a FITC Annexin V Apoptosis Detection Kit (BD Biosciences, Bedford, MA, USA). Then cells were subjected to apoptosis detection by FACSCalibur FCM (BD Biosciences). The data were analyzed by the software FACS Diva (BD Biosciences).
Dual-luciferase reporter system
Plasmid vector psiCHECK2 was purchased from Promega (Madison, WI, USA). 3′UTR sequences of wild-type (WT) and mutant-type (MT) AKAP12 were inserted into the cleavage sites of the psiCHECK2 vector. PsiCHECK2-UTR vectors, negative mimics and miR-338-3p mimics were transfected into cancer cells using Lipofectamine 2000 (Invitrogen). Firefly and Renilla luciferase activities were measured 48 h later using the Dual-Luciferase Reporter System (Promega).
Statistical analysis
Data in this study were analyzed by GraphPad Prism 6.0 (Intuitive Software for Science, San Diego, CA, USA). Values were mean ± standard deviation. Two groups were compared with Student’s t test, while multiple groups were compared using one-way ANOVA. All experiments were independently performed at least three times. The value of p < 0.05 was considered statistically significant.
Results
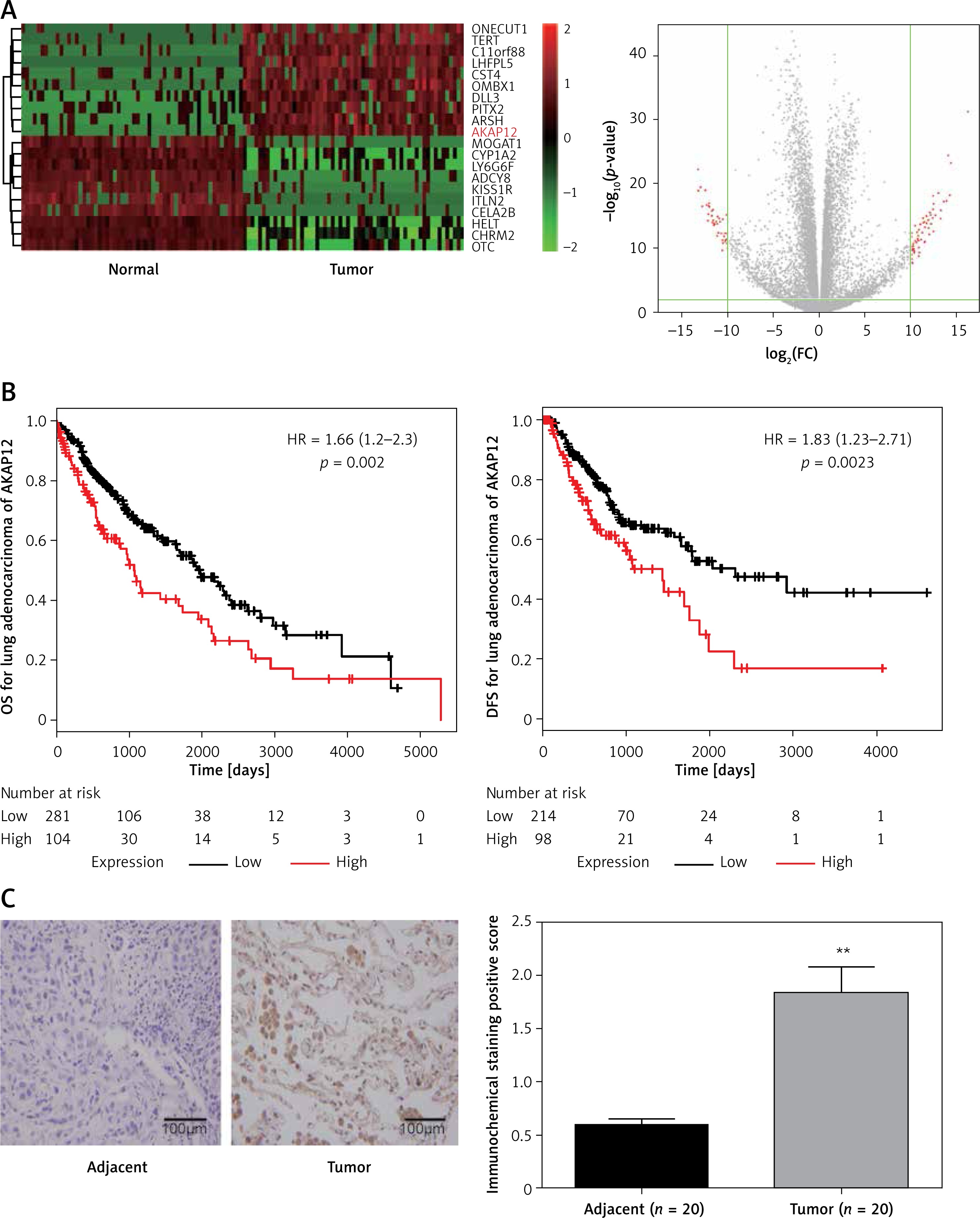
AKAP12 was highly expressed in lung adenocarcinoma
According to the TCGA analysis, 61 genes in total were selected for their aberrant expression in lung adenocarcinoma tissues (n = 58) compared with normal tissues (n = 58). Then 10 high-expressed genes and 10 low-expressed genes were listed in Supplementary Table SI and heatmap and volcano plots were drawn in Figure 1 A. AKAP12, a confusing gene which functioned differently in different cancers, was found overexpressing in lung adenocarcinoma tissues. Analyzed with Kaplan-Meier method, high expression of AKAP12 contributed to worse prognosis regarding shorter disease free survival time (DFS) and overall survival time (OS) (p < 0.01, Figure 1 B). Immunohistochemical staining was performed in 20 paired lung carcinoma tissues and adjacent normal tissues. The staining value for each sample was presented in Supplementary Table SIII and the AKAP12 staining value was higher in tumor tissues (average score = 1.84) compared with normal tissues (average score = 0.60) (Figure 1 C). The aberrantly high expression of AKAP12 could be a potential factor to promote the development of lung adenocarcinoma.
Figure 1
AKAP12 was highly expressed in lung adenocarcinoma tissues. A – Heat map and volcano plot for 20 genes in lung adenocarcinoma. AKAP12 was highly expressed in tumor tissues. B – AKAP12 high expression resulted in worse prognosis. Low expression of AKAP12 significantly lengthened the overall survival time (OS) and disease-free survival time (DFS). C – Immunohistochemistry chart for AKAP12. AKAP12 was highly expressed in tumor tissues compared with adjacent ones regarding immunochemical staining positive score
Up-regulation of AKAP12 deteriorated lung adenocarcinoma in functional experiments
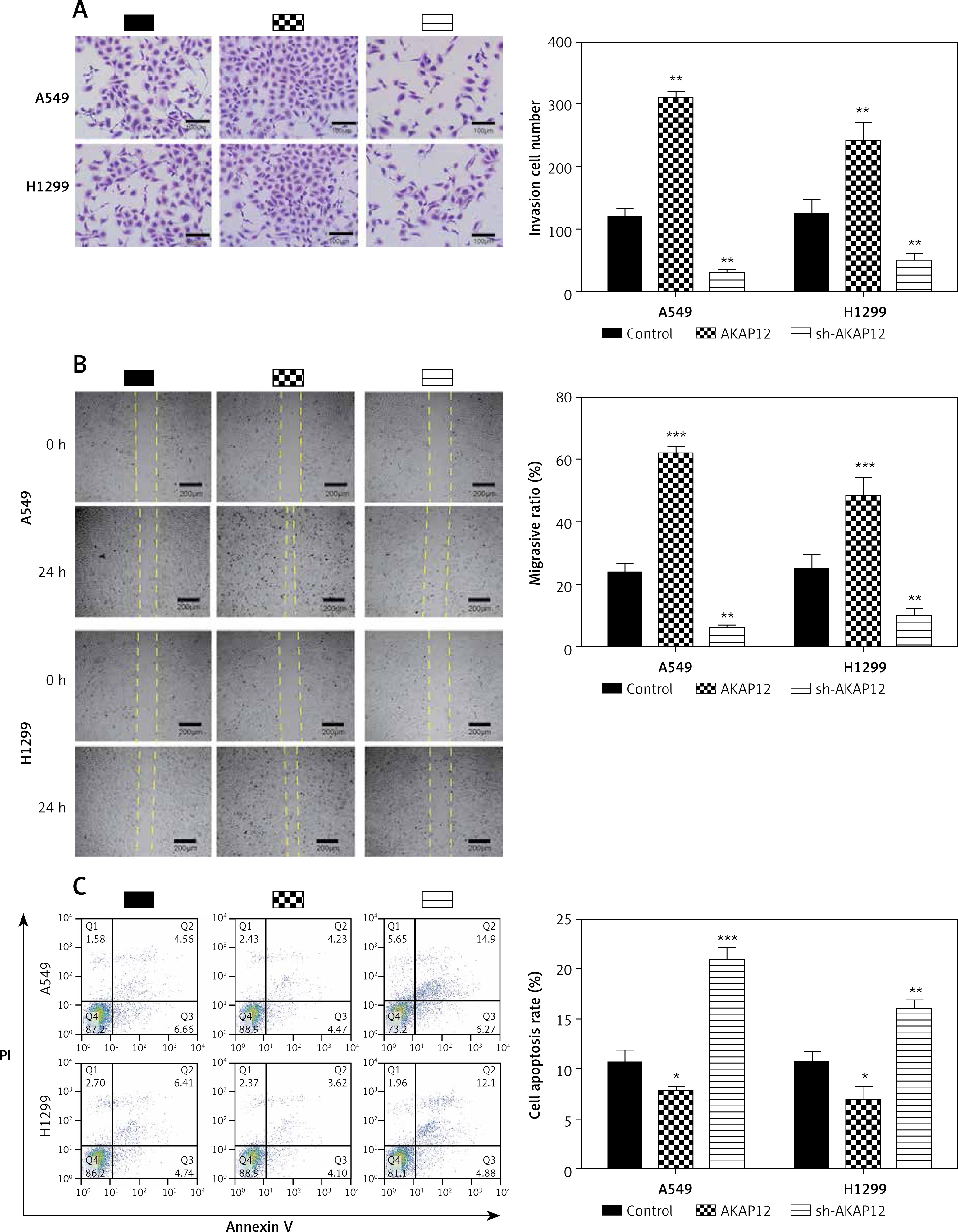
To further investigate the role of AKAP12 in lung adenocarcinoma, we utilized qRT-PCR and Western blot assay and confirmed that both the mRNA and protein level of AKAP12 were higher in cancer cell lines A549 and H1299 compared to the normal cell line HBE (all p < 0.01, Figure 2 A). Transient transfection was performed in both A549 and HBE cells using pcDNA3.1 vector, pcDNA3.1-AKAP12 cDNA or pcDNA3.1-AKAP12 shRNA. At 48 h after transfection, cells were collected and the biological functions were detected. According to the clone formation assay, cell proliferation was significantly upregulated in the AKAP12 group while it was downregulated in the AKAP12 shRNA group (Figure 2 B). Also, cell viability detected by MTT assay in A549 and H1299 cells with overexpression of AKAP12 both displayed an increase (all p < 0.05, Figures 2 C, D). Therefore, AKAP12 overexpression could significantly promote cell proliferation and cell viability, and vice versa. As shown in Figure 3 A, AKAP12 overexpression could dramatically accelerate cell invasion while its suppression could inhibit cell invasion (all p < 0.01). The wound healing assay also displayed a similar trend: at the 24th h after the scratch, more migration appeared in the AKAP12 overexpression group but less migration in the AKAP12 suppression group (p < 0.01, p < 0.001, Figure 3 B). As expected, the cell apoptosis rate was significantly downregulated in the AKAP12 group and was upregulated in the AKAP12 shRNA group, confirming that AKAP12 suppressed cell apoptosis and led to deterioration of lung adenocarcinoma (p < 0.05, p < 0.01, p < 0.001, Figure 3 C).
Figure 2
AKAP12 was highly expressed in lung adenocarcinoma cells. A – AKAP12 was highly expressed in A549 and H1299 cells in protein level. B – AKAP12 was highly expressed in A549 and H1299 cells in mRNA level. C – Clone formation revealed that AKAP12 could promote A549 and H1299 cell proliferation and its knockdown could inhibit proliferation. D – MTT assay revealed that AKAP12 significantly increased cell viability while sh- AKAP12 decreased cell viability
**p < 0.01, compared with control group.
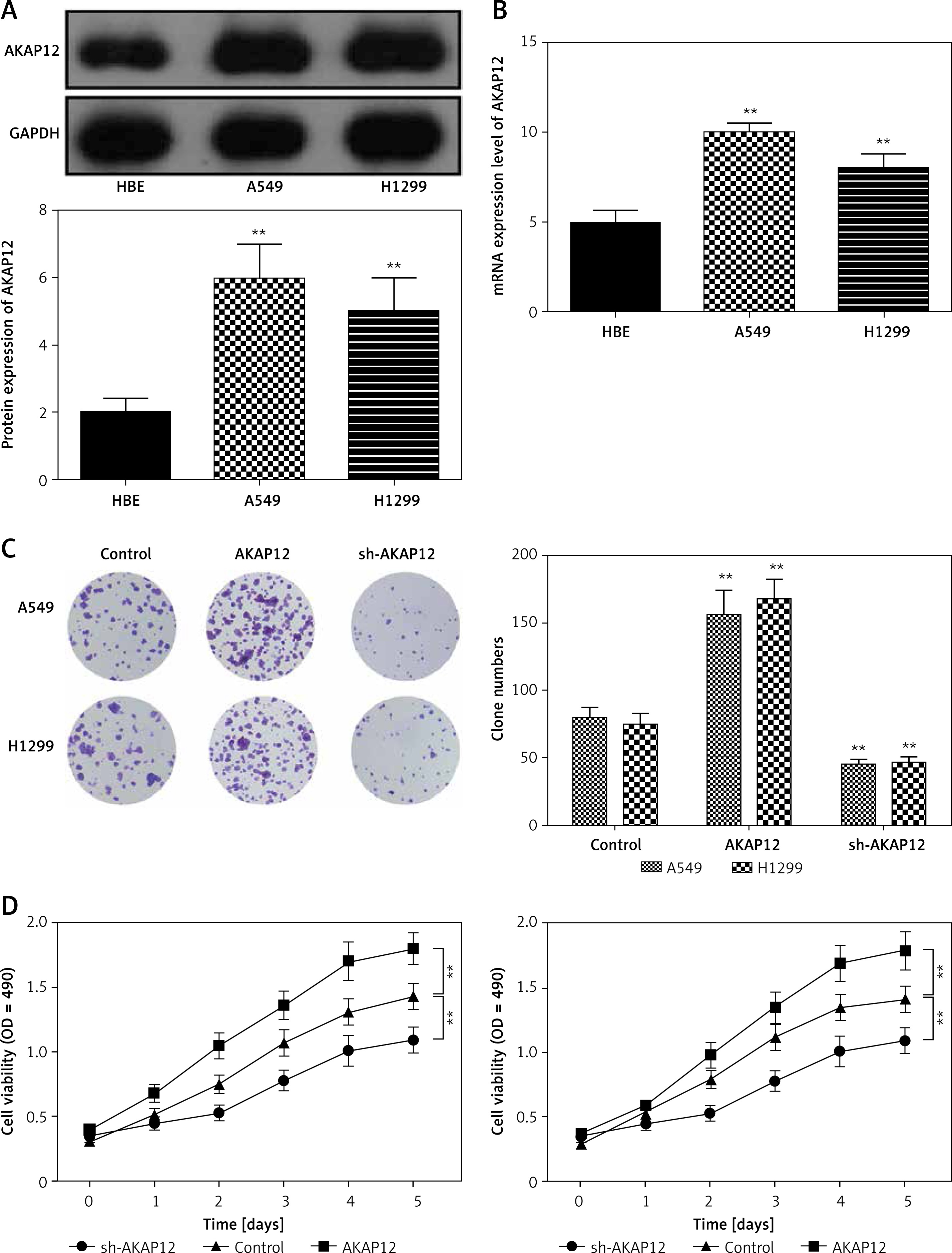
Figure 3
AKAP12 deteriorated lung adenocarcinoma in functional experiments. A – Transwell assay revealed that AKAP12 could increase cell invasion and its knockdown could inhibit the process. B – Wound healing assay revealed that AKAP12 could improve migration ratio and its knockdown could decrease the ratio. C – Flow cytometry revealed that AKAP12 could reduce cell apoptosis rate and its knockdown could increase cell apoptosis rate
*p < 0.05, **p < 0.01, ***p < 0.001, compared with control group.
AKAP12 was the target of miR-338-3p and could be down-regulated by miR-338-3p
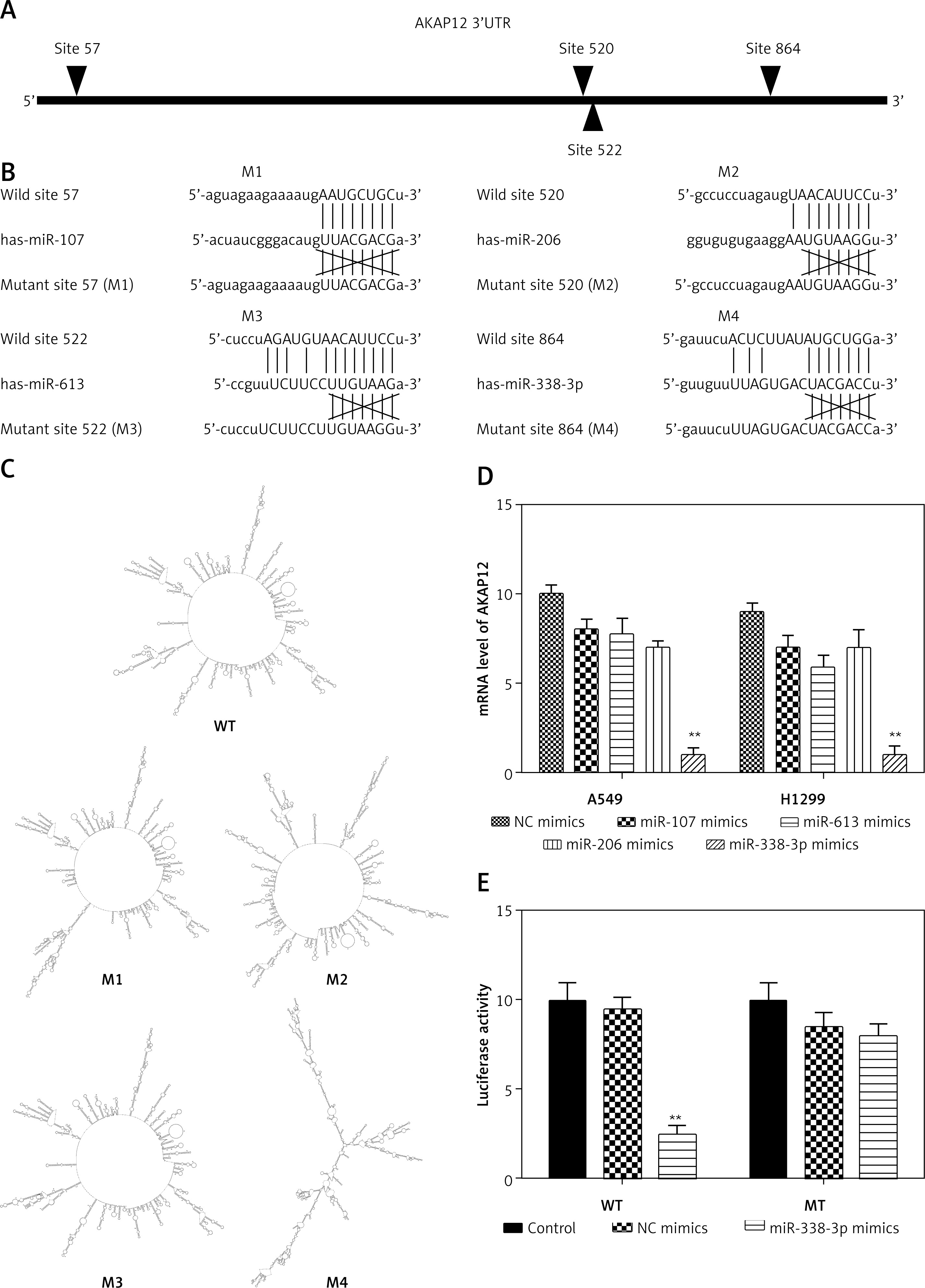
By TargetScan 7.0 and miRNA base selection, four miRNAs significantly regulated AKAP12 in terms of bioinformatics. MiR-107 directly targeted site 57, miR-206 site 520, miR-613 site 522, and miR-338-3p site 864 (Figures 4 A, B). Different effects were judged from mirSVR score, PhastCons score, and context++ score percentile as suggested in Supplementary Table SII. MiR-338-3p had the lowest mirSVR score and a relatively high PhastCons score, indicating the highest thermal stability and a relatively good conservativeness. MiR-338-3p also had the highest context++ score, suggesting the greatest conservation and mRNA destabilization. Therefore, miR-338-3p could be the most suitable candidate for the target gene. Also, AKAP12 structure mutations were predicted according to the RNA Folding Form website, which showed the most significant changes in the miR-338-3p group (Figure 4 C and Supplementary Figures S1–S4). After transfection of 100 nM miRNAs into HEK 293T cells, different effects of four miRNAs on AKAP12 expression were confirmed in Figure 4 D by qRT-PCR detection, and miR-338-3p resulted in the most significant decrease in AKAP12 mRNA expression (both p < 0.01). Scores, structure changes and mRNA level changes of AKAP12 combined confirmed that miR-338-3p could drastically downregulate AKAP12 expression and miR-338-3p was selected as a topic. Then, luciferase activity of the control group, NC mimic group and miR-338-3p mimic group revealed that miR-338-3p directly targeted AKAP12 and downregulated its expression (Figure 4 E).
Figure 4
AKAP12 was the target of miR-338-3p and could be down-regulated by miR-338-3p. A – Targeted sites of four miRNAs are displayed. MiR-107 targeted at site 57, miR-206 at site 520, miR-613 at site 522, and miR-338-3p at site 864. B – Four miRNAs all directly targeted at AKAP12 instead of its variants. C – Structures changes after miRNAs combination according to RNA folding form 23.0. AKAP12 mutant 4 had significant structure changes caused by miR-338-3p. D – Q-PCR of four miRNAs revealed that four miRNAs could downregulate AKAP12 expression, and miR-338-3p displayed the most significant changes. E – MiR-338-3p could inhibit the luciferase activity of WT 3′UTR of AKAP12 but had little influence on MT 3′UTR of AKAP12
**p < 0.01, compared with control group.
MiR-338-3p could inhibit lung adenocarcinoma by AKAP12 suppression
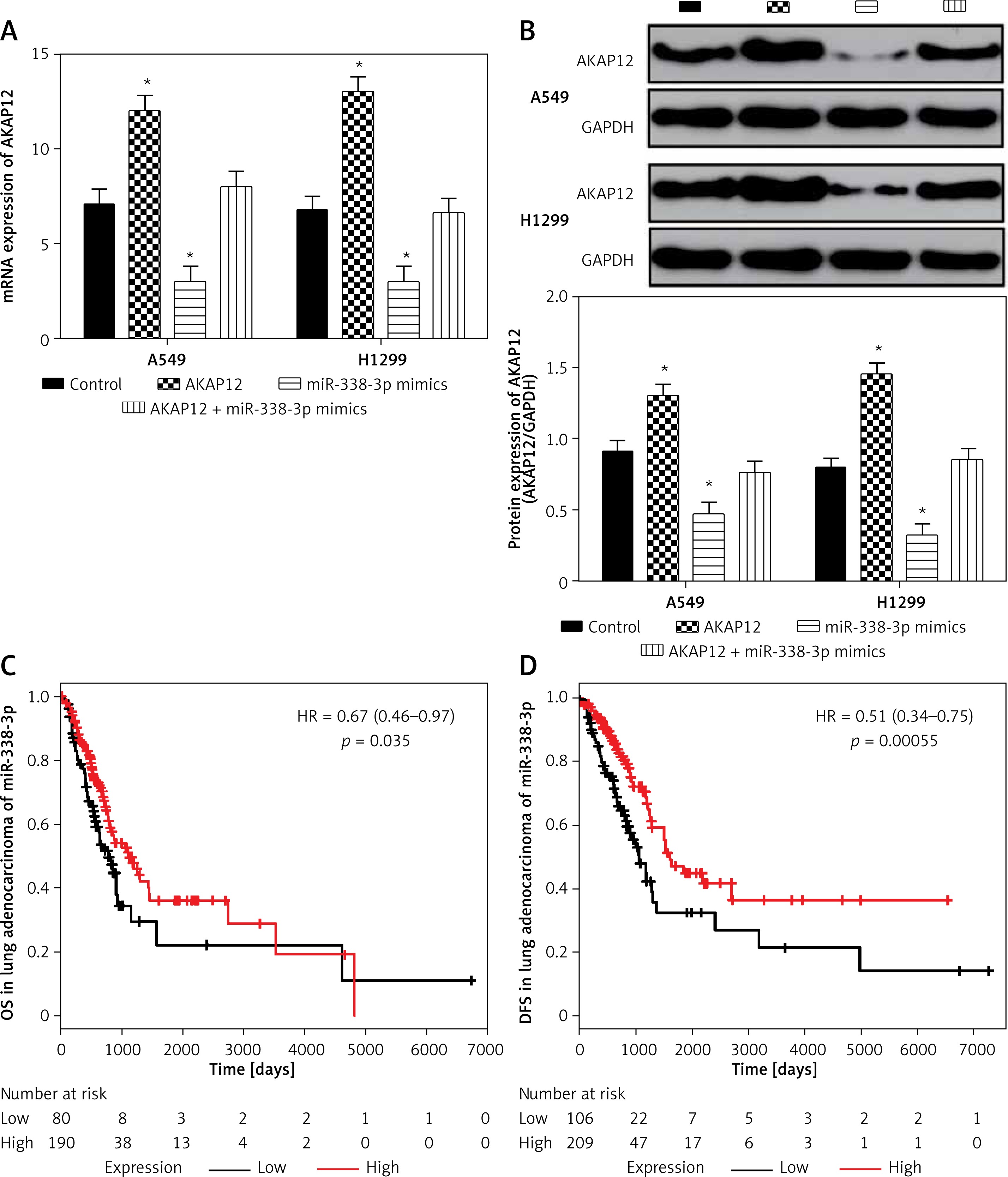
To further understand the effect of miR-338-3p on AKAP12 expression, cells transfected with different plasmids or sequences were divided into four groups, i.e. the control group, AKAP12 group, miR-338-3p mimic group and AKAP12 + miR-338-3p mimic group. AKAP12 significantly upregulated AKAP12 mRNA expression while miR-338-3p downregulated it, but AKAP12 and miR-338-3p combined caused no significant differences (p < 0.05, Figure 5 A). AKAP12 protein expression was upregulated in the AKAP12 group and downregulated in the miR-338-3p group, but showed no significant differences in the AKAP12 + miR-338-3p mimic group (p < 0.05, Figure 5 B). Also, after dividing the patients into two groups according to the median miR-338-3p expression, we found that high expression of miR-338-3p would improve the prognosis, considering the higher DFS and OS rates (Figures 5 C, D). With the Kaplan-Meier method and Cox regression models, univariate and multivariate analysis for the overall survival of lung adenocarcinoma was performed using the data from the TCGA database (Tables II and III). Stage event pathologic stage (I–IV), M stage, miR-388-3p and AKAP12 expression were all correlated with the prognosis in univariate analysis while both miR-388-3p and AKAP12 expression were recognized as independent prognostic factors for the overall survival. The above results verified that miR-338-3p suppressed the progression of lung adenocarcinoma by inhibiting AKAP12.
Table II
Univariate analysis for overall survival
Discussion
Our study indicated that the expression level of AKAP12 was high in lung adenocarcinoma. Meanwhile, high expression of AKAP12 predicted a poor prognosis. AKAP12 alpha previously was reported to be a promoter in lung cancer in the research of Jo et al.; AKAP12α protein was downregulated in H358, H460 and Calu-1 cells but not H1299 cells, which to a degree is consistent with this study. The methylation status of 5′CpG islands of the AKAP12α gene in lung tissues might be the reason for the promotion [17]. AKAP12, which was methylated at nearly 40% prevalence in lung adenocarcinoma samples and modulates the expression of genes involved in the cell cycle and apoptosis, was not associated with age at diagnosis or stage of lung cancer [22]. This study by contrast proved a relationship between AKAP12 and DFS as well as OS, which hallmarked its biomarker role in lung adenocarcinoma prognosis. After manipulating the expression of AKAP12 in A549 and H1299 cells, we confirmed that AKAP12 acts as an oncogene and facilitated the proliferation, migration and invasion of lung cancer cells.
The promotion effect of AKAP12 on lung adenocarcinoma was associated with miR-338-3p as we proved the target relationship between miR-338-3p and AKAP12. MiR-338 was reported to abate lung cancer in vitro and in vivo by targeting ITGB3 [13], a conclusion similar to this study. MiR-338-3p could suppress lung cancer tumorigenesis and metastasis in vitro and in vivo by targeting the EMT regulator Sox4 as well [9]. MiR-338-3p was also predicted in an integrative bioinformatics analysis as a biomarker for diagnosis of NSCLC adenocarcinoma [23]. In this research, miR-338-3p was discovered to target AKAP12 and inhibited lung adenocarcinoma development, which to our knowledge had not been studied before. As a result, our study demonstrated a new regulatory mechanism of miR-338-3p, which revealed a better strategy for lung adenocarcinoma treatment.
However, there are some limitations in this study. The underlying mechanisms of AKAP12 regulation in lung adenocarcinoma remains to be further investigated including methylation and phosphorylation. AKAP12α and AKAP12β expression and corresponding results still need exploration as well as in the case of opposite results when it comes to lung adenocarcinoma. MiRNAs which may have better combination with AKAP12 could be discussed in future studies for the consideration of better application in lung adenocarcinoma treatment.
In conclusion, AKAP12 was upregulated in lung adenocarcinoma, and its high expression promoted tumor development and predicted poor prognosis. High expression of AKAP12 in lung adenocarcinoma led to proliferation, migration and invasion of cancer cells, and apoptosis inhibition. MiR-338-3p directly targeted AKAP12 and inhibited lung adenocarcinoma by AKAP12 restraint. The regulation of miR-338-3p and AKAP12 could be of value for clinic targeted therapy of lung adenocarcinoma.