
Chronic myelogenous leukemia (CML) is characterized by a t(9;22) chromosome translocation (Philadelphia chromosome-positive [Ph+]) that creates the BCR/ABL oncogene. This fusion protein displays constitutive tyrosine kinase activity, leading to the induction of aberrant proliferation and neoplastic transformation [1]. The Ph+ chromosome is found in more than 95% of CML and in Ph+ acute lymphoblastic leukemia.
ABL tyrosine kinase inhibitors (AKIs) are utilized for the treatment of Ph+ leukemia; the initial response is beneficial [2–4] but unfortunately, the clinical efficacy of this treatment decreases steadily as the disease progresses [5]. CML blast crisis or Ph+ acute lymphoblastic leukemia patients only benefit from TKI treatment temporarily, if at all [6, 7].
Resistance to drug therapy in Ph+ leukemia is mediated by mutations within BCR/ABL or by BCR/ABL-independent mechanisms, such as tumor microenvironment-mediated drug resistance [8]. The bone marrow (BM) plays a vital role in hematopoiesis, as well as in different aspects of disease progression in hematological malignancies [8, 9]. The BM microenvironment is also critical for long-term hematopoiesis, and for the maintenance and regulation of stem cells and their progeny [9]. It is a rich source of paracrine- and autocrine-derived factors which have also been implicated in drug resistance for both hematologic malignancies and solid tumors that metastasize to the BM [10–12]. Conditions within the BM niche, including soluble factors (SFs), interleukins (ILs), stromal cells, and extracellular components, contribute to reduced drug sensitivity of cancer cells [10, 13, 14], including drug resistance to multiple TKIs, such as imatinib, nilotinib, and dasatinib [15–19].
To explore the BCR/ABL-independent mechanisms underlying Ph+ leukemia drug resistance, we investigated the ability of SFs collected from mesenchymal stem cells (MSCs) to confer drug resistance in CML cell lines in vitro. Exposure of CML cells to mesenchymal SFs conferred resistance to imatinib, but not crizotinib, which was mediated in part by the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway. Inhibition of JAK2 by ruxolitinib restored sensitivity to imatinib in CML cells exposed to mesenchymal SFs. Moreover, the multi-TKI crizotinib, which has been reported to also inhibit ABL tyrosine kinase activity, was also able to abrogate JAK2 activity, thereby overcoming SF-mediated drug resistance in Ph+ leukemia.
Blast crisis human chronic myelogenous leukemia cell line K562 and megakaryocytic leukemia cell line MEG-01 (American Type Culture Collection (ATCC), VA, USA), and Ba/F3 JAK2 V617F cells expressing activated JAK2 and harboring the V617F mutation, have been described previously [20]. Cells were cultured in RPMI 1640 complete medium supplemented with 10% (w/v) fetal bovine serum, 1% (w/v) L-glutamine, and penicillin/streptomycin. The human and murine stromal cells HS-5 and MS-5, respectively (ATCC), were maintained under the same conditions. All cells were grown at 37°C in a humidified atmosphere with 5% CO2.
HS-5 or MS-5 cells were grown in complete RPMI medium and CM were collected after 72 h. Contaminating cells were cleared by centrifugation at 1,000 rpm for 1 min; the collected supernatant was used fresh in each experiment.
Cells (2 × 105/well) were plated in six-well plates. After 24 h, cells were treated with the specified agents. Solvent-treated samples were incubated with 1% (w/v) dimethyl sulfoxide. Cells were collected 72 h later, stained with 0.4% (w/v) trypan blue solution (1 : 1, v/v), and counted using a hemacytometer [20].
Cytokine levels were measured in the CM of MS-5 cells using mouse cytokine array panel A (Proteome Profiler Array, R&D Systems, MN, USA). Briefly, CM were mixed with a cocktail of biotinylated detection antibodies. The CM–antibody mixture was then incubated with the mouse cytokine array membrane. Following a wash to remove unbound material, streptavidin conjugated to horseradish peroxidase and chemiluminescent detection reagents were added sequentially. Data were visualized using X-ray film. Optical densities were determined using ImageJ software.
Clonogenicity assay was performed as previously described [21]. The plates were stained for 4 h with 5 mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, and the dye was extracted with solubilization buffer (20% w/v sodium dodecyl sulfate (SDS), 50% w/v N,N-dimethylformamide, 25 mM HCl) for 24 h. Optical density was measured at 570 nm with a reference wavelength of 630 nm.
Immunoblotting was performed as described previously [22]. Anti-pSTATs (phosphor-Stat antibody sampler kit cat #9914), anti-JAK2 (cat #3230), anti-pJAK2 (Tyr 1007/1008)(cat #3776), anti-phosphor Abl (Tyr 245) (cat #2861) and anti-cleaved PARP (cat #5625) antibodies were from Cell Signaling Technology (MA, USA). Anti-PARP (cat #SC-8007) antibodies were from Santa Cruz Biotechnology (CA, USA).
Student’s t-test was used for the statistical analyses, with the significance level set at p < 0.01 or p < 0.001.
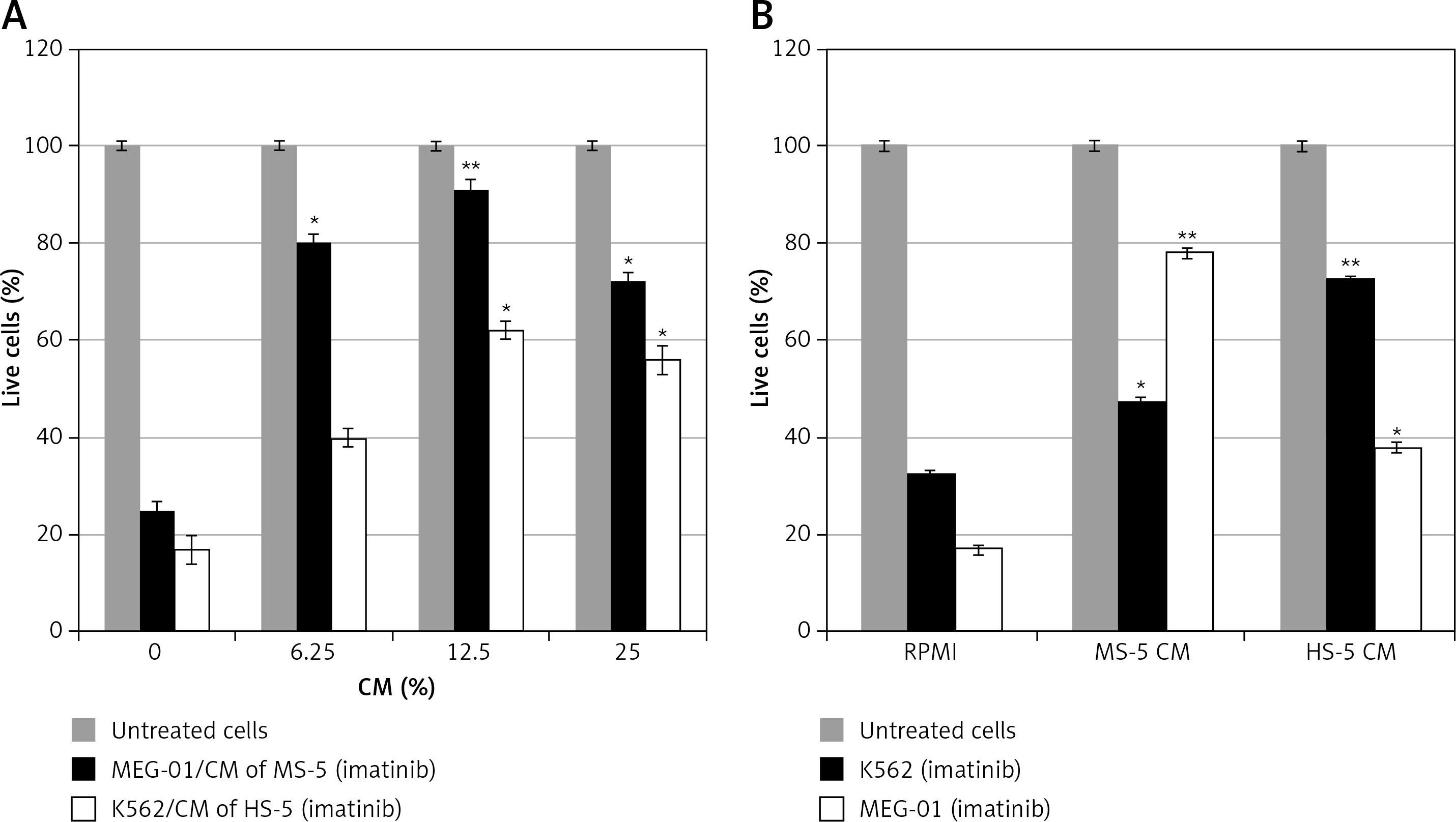
To address drug sensitivity of CML cell lines to AKIs in the presence of CM collected from MSC cells, we utilized human K562 [23] and MEG-01 cells [24] as an in-vitro model of CML. We followed the effect of imatinib on proliferation and triggering of PARP cleavage as a marker of apoptosis induction [25] in MEG-01 CML cells in the presence and absence of CM collected from MS-5 cells. Exposure of MEG-01 cells to various concentrations of MS-5 CM (6.25–25%) resulted in increased cell survival in the presence of imatinib (Figure 1 A). In fact, as little as 3% MS-5 CM was sufficient to confer partial drug resistance in MEG-01 cells treated with imatinib (data not shown). Next, we measured the effect of MS-5 CM on imatinib-induced growth inhibition using K562 and MEG-01 CML cells. MS-5 CM protected cells of both CML lines from imatinib-induced growth inhibition, albeit with varying potency. Overall, the magnitude of the drug resistance observed in MEG-01 cells was more than 2-fold greater than that in K562 cells (Figure 1 B). This suggested that murine SFs are active against human CML cells, although the basis for the different responses of K562 and MEG-01 cells to MS-5 CM is unclear. Nevertheless, we chose to further focus on drug sensitivity of MEG-01 cells in response to exposure to MS-5 CM.
Figure 1
Mesenchymal stem cell (MSC) conditioned medium (CM) confers drug resistance to imatinib in CML cells. A – MEG-01 and K562 cells were exposed to increasing concentrations of CM (0, 6.25, 12.5, 25%) collected from MS-5 or HS-5, and viability of remaining cells after treatment with 1 μM imatinib for 72 h was determined. CM collected from MS-5 or HS-5 supplied to MEG-01 and K562, respectively. B – MEG-01 and K562 cells were exposed to 12.5% CM collected from MSC cells in the presence or absence of 1 μM imatinib for 72 h. Negative control – 12.5% RPMI medium +1% FCS. *,**Significantly different from untreated cells at p < 0.01 and 0.001, respectively. The experiment was repeated twice, with a comparable outcome
We asked whether CM collected from other cells might promote resistance to imatinib in MEG-01 cells. CM collected from A2780 and B16-F10 cell lines were not efficient at conferring drug resistance to imatinib (Supplementary Figure S1 A). Additional data (Supplementary Figure S1 B) supported involvement of SFs, but not microvessels, as mediators of drug resistance in CML exposed to MSC CM [26].
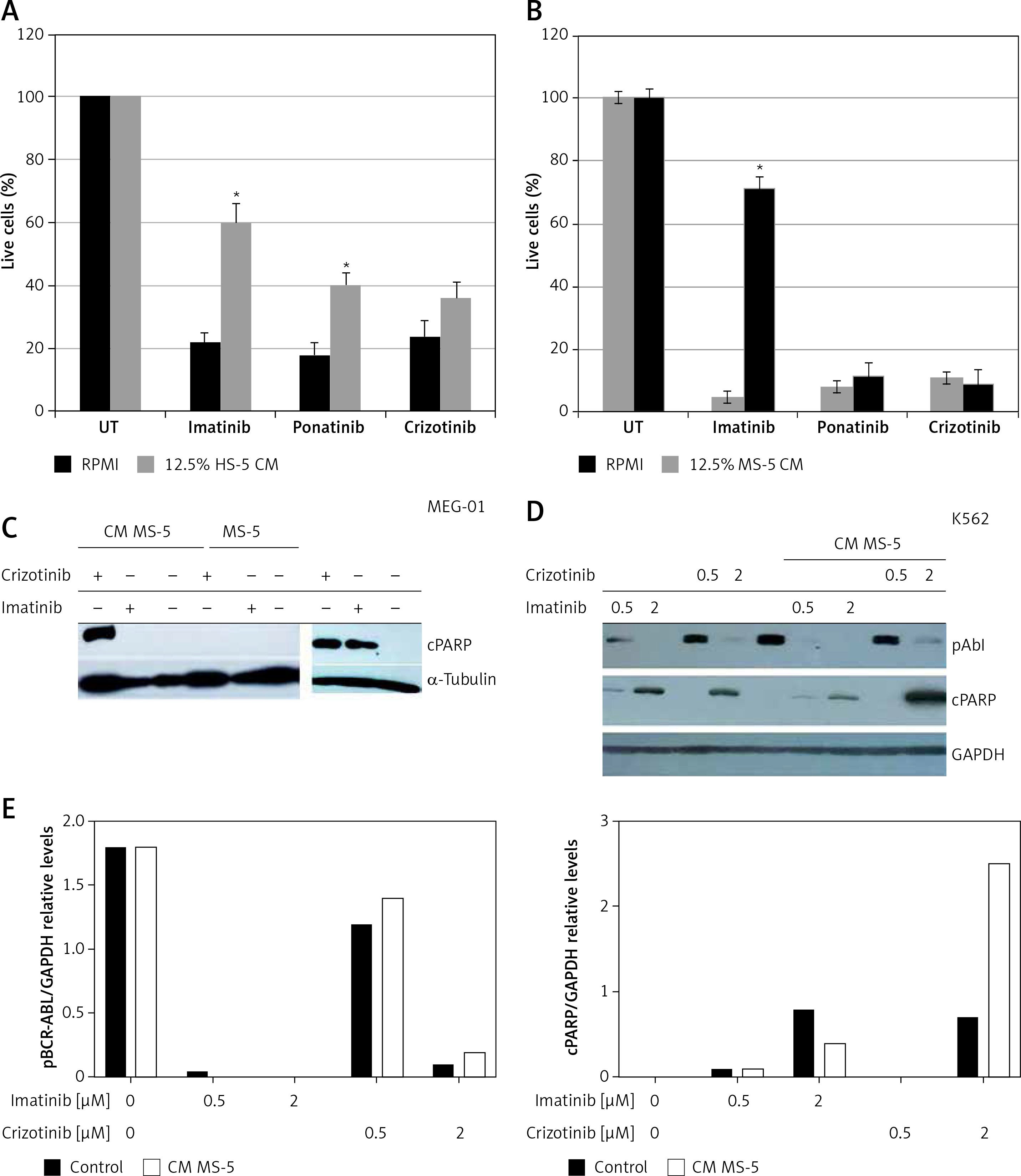
We recently demonstrated that crizotinib, an anaplastic lymphoma kinase (ALK)/ROS1 inhibitor [27, 28], efficiently and selectively suppresses growth of Ph+ cells, and inhibits activity of native and T315I-mutated BCR/ABL [29, 30]. Here, we also explored the ability of CM collected from MSCs to induce drug resistance in MEG-01 cells exposed to crizotinib. Figure 2 shows that MS-5 CM conferred significant drug resistance to imatinib in MEG-01 cells, but had a minimal protective effect in cells exposed to either ponatinib [31] or crizotinib (Figure 2 A). Next, we monitored the ability of MSC CM to interfere with imatinib- and crizotinib-induced apoptosis of MEG-01 cells. Initially, MEG-01 cells were treated with crizotinib or imatinib for 24 h in the presence or absence of MS-5 CM. Exposure to imatinib/crizotinib significantly increased the levels of cleaved PARP enzyme (Figure 2 B) [25]. Interestingly, the presence of MS-5 CM prevented PARP cleavage, again suggesting that it confers drug resistance to imatinib in MEG-01 cells (Figure 2 B). In contrast, exposure of MEG-01 cells to crizotinib resulted in significant cleavage of PARP, indicating induction of apoptosis (Figure 2 B). Next, we examined the effect of MS-5 CM on the activity of BCR/ABL in MEG-01 cells exposed to imatinib or crizotinib. Data presented in Figures 2 C and D show that imatinib was active in inhibiting BCR/ABL auto-phosphorylation in the presence and absence of MS-5 CM. In addition, the high concentration of crizotinib was active at inhibiting BCR/ABL auto-phosphorylation, but showed only marginal activity at the low concentration, in the presence and absence of MS-5 CM. Imatinib was active at inducing PARP cleavage in MEG-01 cells, and induction of PARP cleavage was largely reduced (by about 50%) in the presence of MS-5 CM. In contrast, crizotinib induction of PARP cleavage was not reduced, but rather enhanced in the presence of MS-5 CM. Thus, the apoptosis-inducing activity of imatinib, but not of crizotinib, was reduced in the presence of MS-5 CM with no significant change in imatinib’s ability to inhibit BCR/ABL auto-phosphorylation.
Figure 2
Crizotinib overcomes mesenchymal stem cell (MSC) conditioned medium (CM)-mediated drug resistance. A – MEG-01 cells were exposed to 1 µM imatinib, ponatinib or crizotinib in the presence or absence of 12.5% HS-5 CM and the number of viable cells was counted after 72 h exposure. B – MEG-01 cells were exposed to 1 µM imatinib, ponatinib or crizotinib in the presence or absence of 12.5% MS-5 CM and the number of viable cells was counted after 72 h exposure. C – Immunoblot of MEG-01 cells exposed to 1 µM imatinib or crizotinib in the presence or absence of MS-5 CM. In addition, 1 mM imatinib or crizotinib was added to MEG-01 cells co-cultured with MS-5 cells. Filters were probed with anti-cleaved (c)-PARP antibody and a-tubulin was used as a loading control. D – Immunoblot of K562 cells exposed to 0.5 µM and 2 µM imatinib or crizotinib in the presence or absence of MS-5 CM. Filters were probed with anti-c-PARP antibody, anti-phospho Abl (Tyr 245) and anti-GAPDH was used as a loading control. E – Quantitation of experiment in D showing relative levels of cleaved PARP and phospho BCR/ABL (relative to GAPDH). *Significantly different from untreated cells at p < 0.01. The experiment was repeated twice, with a comparable outcome
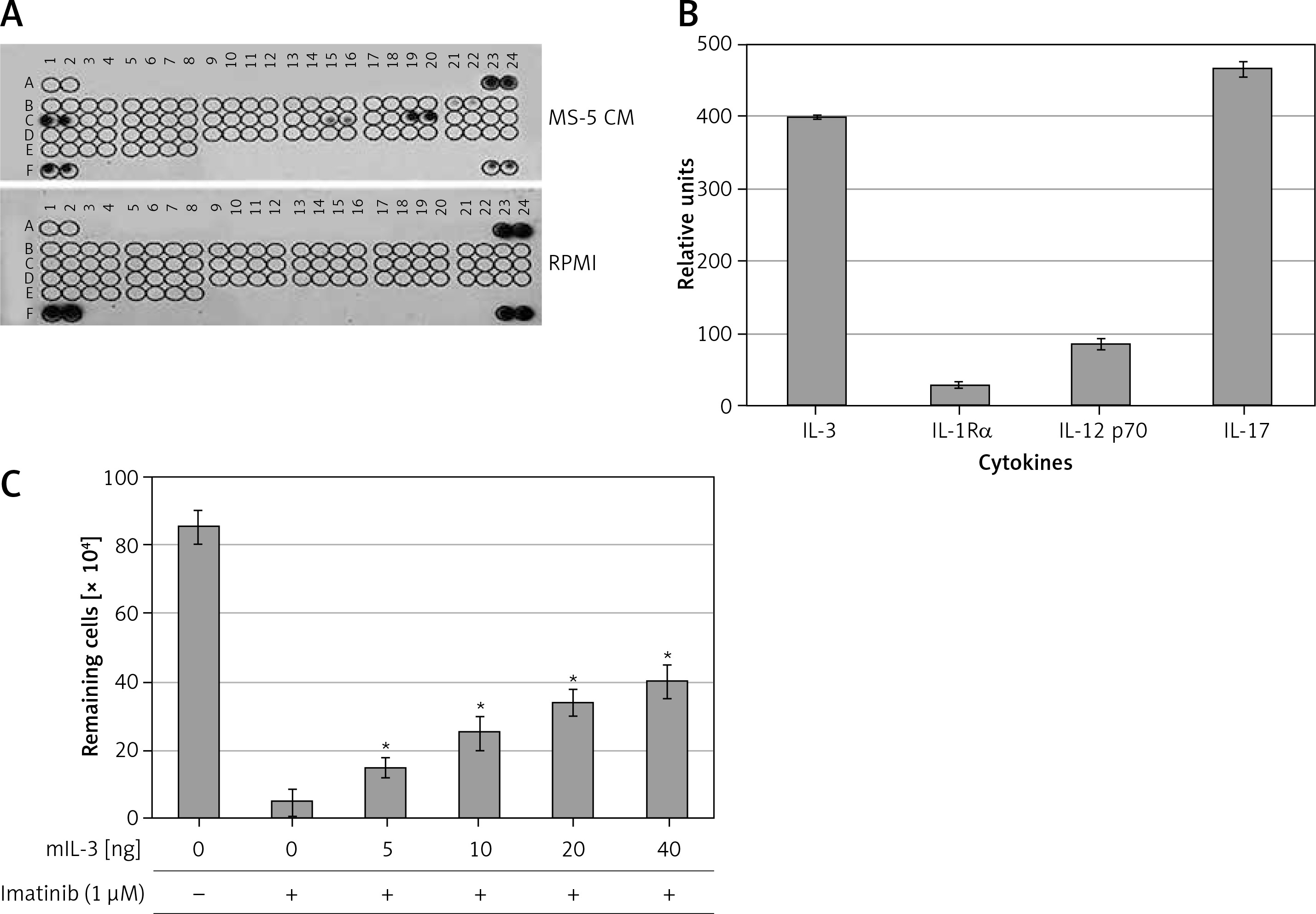
To identify potential SFs in the MS-5 CM that are involved in mediating drug resistance to imatinib, we monitored the levels of a variety of SFs using a cytokine array assay kit. Figures 3 A and B shows a number of cytokines and ILs, in particular IL-3 and IL-17 (Figures 3 A, B), that were present at high levels in the MS-5 CM, whereas others, such as IL-1Rα and IL-12 p70, were present at low levels (Figures 3 A, B). We hypothesized that one or a few cytokines are involved in mediating imatinib drug resistance in CML cells, and we selected IL-3 to further investigate this. Increasing concentrations of murine IL-3 conferred resistance to imatinib in MEG-01 cells in a dose-dependent manner (Figure 3 C).
Figure 3
Identification of soluble factors (SFs) in MS-5 conditioned medium (CM) as potential mediators of imatinib resistance in MEG-01 cells. CM was collected from MS-5 cells after 72 h of culture. A – Immunoblot test for relative expression levels of 40 factors in MS-5 CM was carried out according to the manufacturer’s instructions. B – Optic absorption was measured by TINA software and levels of soluble factors in MS-5 CM relative to RPMI are presented. C – Cell viability (number of remaining cells) of K562 cells treated with 1 µM imatinib and increasing concentrations of murine (m) IL-3 for 72 h was determined. *Significantly different from untreated cells at p < 0.01. The experiment was repeated twice, with a comparable outcome
JAK/STAT pathways are downstream regulators of a number of ILs, such as IL-6 and IL-3, which have also been implicated in drug resistance in CML [15, 32, 33]. Moreover, emerging data also implicate the JAK/STAT pathway in BM-mediated drug resistance [15, 16, 19]. Thus, we investigated the involvement of the JAK/STAT pathway in the observed imatinib resistance in CML cells exposed to MS-5 CM. MEG-01 cells were exposed to MSC CM and levels of various phosphorylated STATs were monitored.
The levels of both pSTAT1 and pSTAT2 were undetectable in the MEG-01 cells, with no change after exposure to either MS-5 CM (Figure 4 A) or HS-5 CM (data not shown). Moreover, STAT6 and STAT3 were not phosphorylated in MEG-01 cells, and exposure to MS-5 or HS-5 CM significantly stimulated their phosphorylation levels. In contrast, STAT5, which is a direct downstream target of BCR/ABL [34–36], was significantly phosphorylated in MEG-01 cells and the phosphorylation levels were further increased upon exposure to both HS-5 and MS-5 CM (Figure 4 A).
Figure 4
MS-5 conditioned medium (CM) stimulates JAK/STAT pathway in MEG-01 cells. A – MEG-01 cells exposed to 12.5% CM collected from MS-5 and HS-5 cultures were used to monitor levels of activated STATs by immunoblotting. MEG-01 cells were seeded and exposed to 12.5% CM from MS-5/HS-5 cells for 2 h. Cells were collected, and the phosphorylation of STAT3/4/6 was monitored by immunoblotting. Tubulin was used as a loading control. B – MEG-01 cell viability was monitored after exposure to 12.5% MS-5 CM in the presence of 1 μM imatinib, imatinib + ruxolitinib or crizotinib alone. Cell viability was monitored 72 h after treatment. C – Ability of different kinase inhibitors to induce apoptosis in MEG-01 cells in the presence of MS-5 CM. MEG-01 cells were treated with 1 µM of the different Abl kinase inhibitors in the presence or absence of 12.5% CM for 24 h. The complete PARP protein (120 kDa) was used as a positive control. *,**Significantly different from untreated cells at p < 0.01 and 0.001, respectively. The experiment was repeated twice, with a comparable outcome
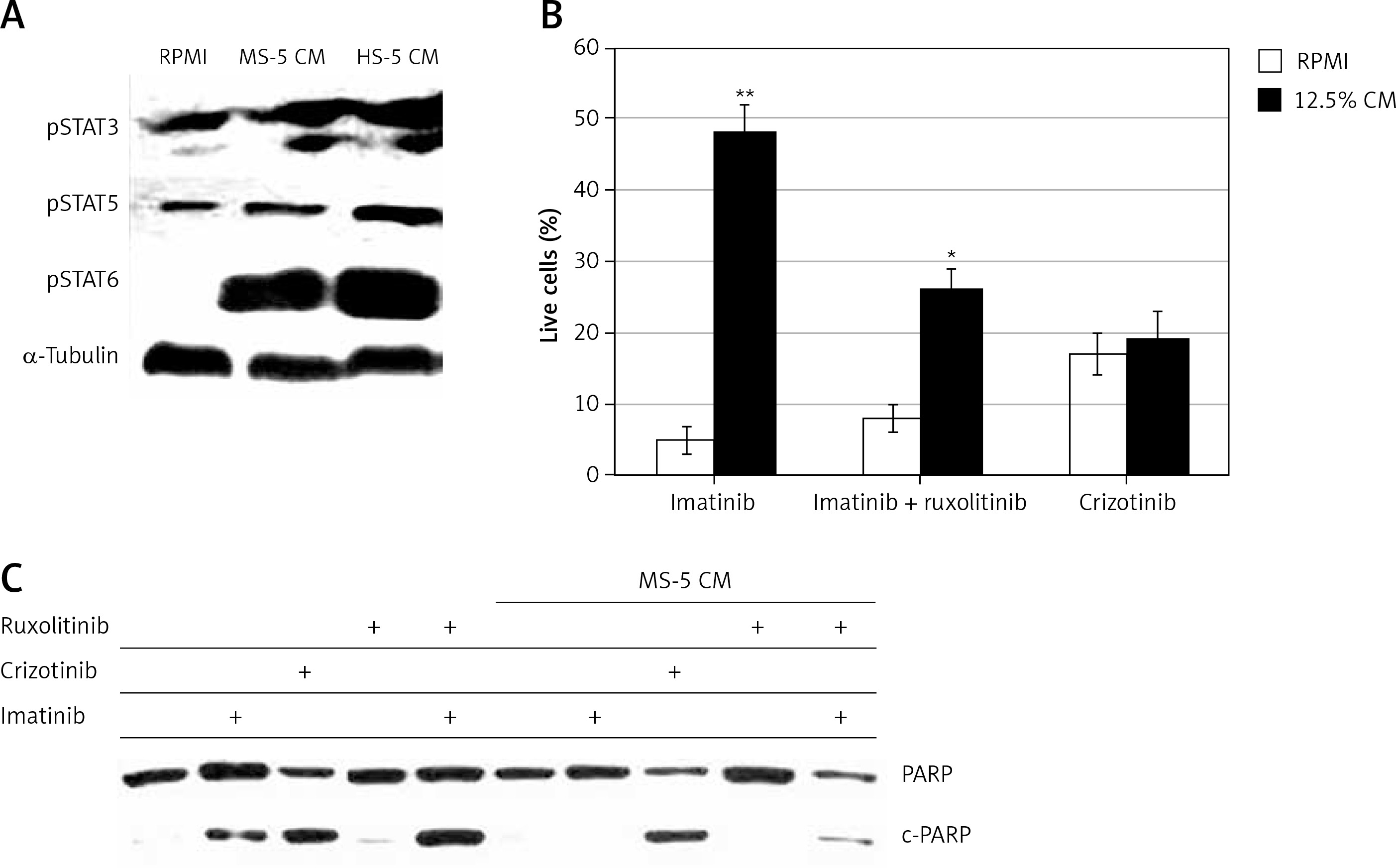
STATs are downstream transcription factors that are activated by JAKs in response to cytokine activation. Thus, we investigated the influence of JAK inhibitors on imatinib resistance mediated by MS-5 CM. Figure 4 B shows the viability of MEG-01 cells exposed to MS-5 CM in the presence of imatinib and the clinically approved JAK1/2 inhibitor ruxolitinib [37]. As expected, exposure of MEG-01 cells to MS-5 CM conferred drug resistance to imatinib, but not crizotinib. When ruxolitinib was added along with imatinib, MEG-01 sensitivity to the latter was partially restored (Figure 4 B), supporting the involvement of JAK1/2 in mediating the MSC CM-induced drug resistance. Further support for the ability of ruxolitinib to restore sensitivity to imatinib in the presence of MS-5 CM was provided by following the levels of cleaved PARP in MEG-01 cells treated with imatinib, crizotinib, ruxolitinib, or imatinib + ruxolitinib in the presence and absence of MS-5 CM. Imatinib and crizotinib induced PARP cleavage in MEG-01 cells. However, no PARP cleavage was observed in MEG-01 cells treated with either imatinib or ruxolitinib alone in the presence of MS-5 CM (Figure 4 C). On the other hand, ruxolitinib + imatinib effectively caused significant PARP cleavage, indicating restoration of partial sensitivity of MEG-01 cells to imatinib in the presence of MS-5 CM (Figure 4 C).
Data presented in Figures 4 B and C show that inhibition of JAK1/2 (by ruxolitinib) can restore sensitivity to imatinib in CML cells exposed to MS-5 CM. We therefore speculated that crizotinib’s ability to overcome mesenchymal SF-mediated drug resistance in CML might be related to JAK1/2 activity. To address this point, we utilized a Ba/F3 model system in which cells become IL-3-independent upon introduction of activated JAK2 (JAK2 V617F) [20]. Use of this system allowed us to monitor the direct effect of crizotinib on JAK2 activity. Initially, we monitored the effects of crizotinib and other relevant TKIs on the clonogenicity of Ba/F3 JAK2 V617F cells. Figures 5 A and B shows that the colony-forming ability of Ba/F3 JAK2 V617F cells was not affected by imatinib or other ABL inhibitors such as GNF-2 or GNF-5 [38]. However, crizotinib inhibited clonogenicity of Ba/F3 JAK2 V617F cells with a potency comparable to that of ruxolitinib. Interestingly, ponatinib was also a potent inhibitor of Ba/F3 JAK2 V617F clonogenicity.
Figure 5
Crizotinib inhibits JAK2 activity. A – Ba/F3 JAK2 V617F cells were used to monitor different kinase inhibitors’ effects on clonogenicity as described in Material and methods. B – Areas of each sample were determined by monitoring absorbance using Image J and values obtained in samples treated with 0.1 µM and 1 µM kinase inhibitor were plotted. C – Ba/F3 JAK2 V617F cells were exposed for 1 h to different concentrations of imatinib, crizotinib (1, 3 and 10 µM) or GNF-5 (3 and 10 µM). Ruxolitinib at 1 µM was used as a positive control. Cells were collected and lysed and their proteins were separated by SDS-PAGE. Levels of pJAK2 were monitored relative to JAK2 and a-tubulin was used as a loading control. D – Levels of pJAK2 relative to a-tubulin. The experiment was repeated twice, with a comparable outcome
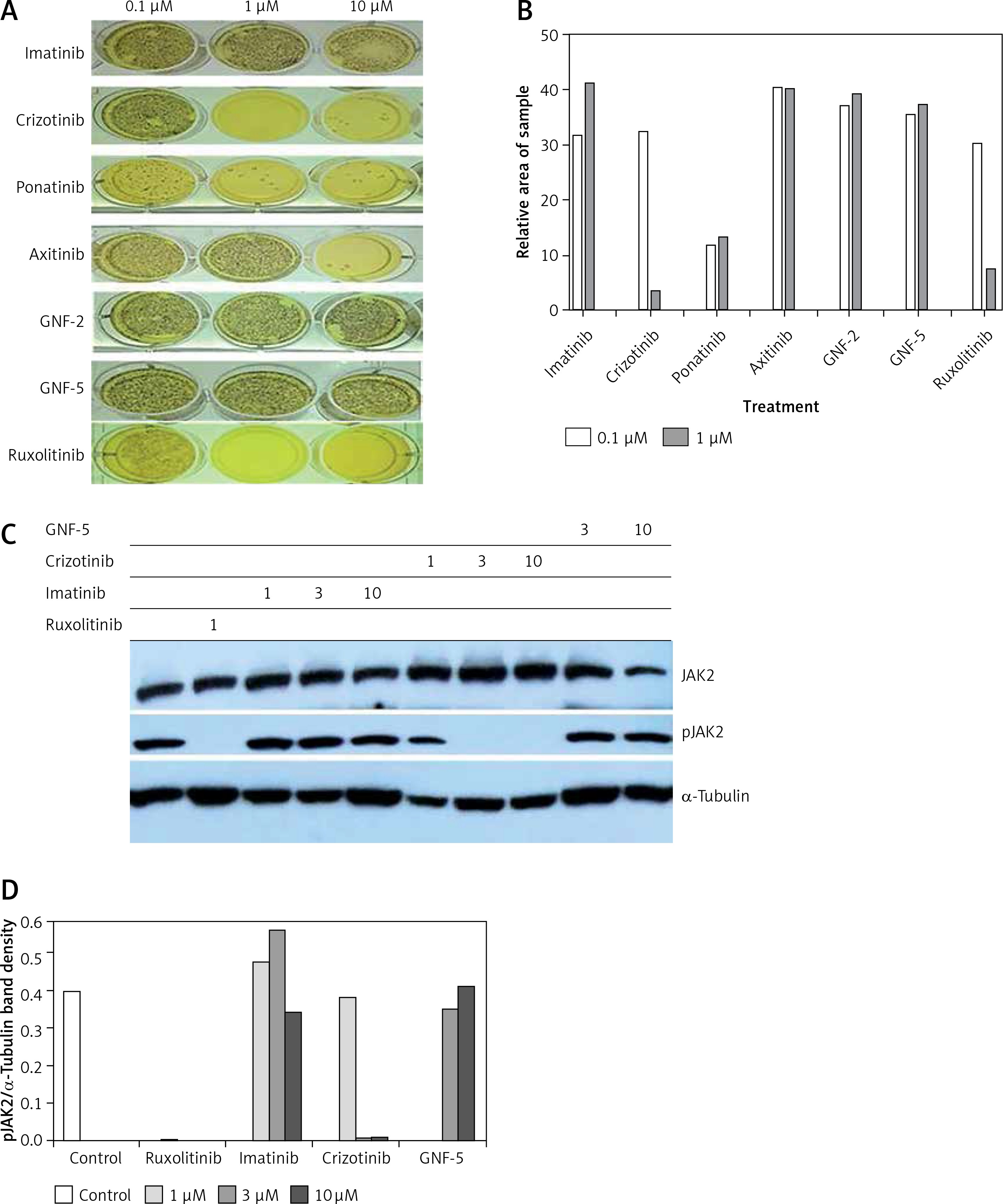
Next, we utilized the same cells to monitor direct inhibition of JAK2 auto-phosphorylation by crizotinib. Ruxolitinib actively inhibited auto-phosphorylation of JAK2 V617F, and crizotinib also exhibited strong inhibition of JAK2 auto-phosphorylation. In contrast, neither imatinib nor GNF-5 inhibited JAK2 auto-phosphorylation (Figures 5 C, D).
CML-targeted therapy has proven to be very effective in managing the disease and has led to extended lifespans for many CML patients. However, about 30% of those patients fail to respond, respond suboptimally, or experience disease relapse after treatment with CML-targeted therapy due, in part, to drug resistance [5, 39]. In general, Ph+ leukemia resistance to drug therapy might be due to mutations in the cancer cells, including alterations in the BCR/ABL fusion gene, or associated with interactions between Ph+ leukemia cancer cells and the BM microenvironment, leading to tumor microenvironment-mediated drug resistance and consequently a low level of residual disease and disease progression [8].
Intensive effort has been invested in developing strategies to address drug resistance in Ph+ leukemia by introducing new TKIs that are active against ABL1 mutations, including the gatekeeper T315I mutation [40]. Moreover, effort is still being made to target additional regulatory elements within the ABL1 kinase, such as the myristoyl-binding pocket in the BCR/ABL protein [20, 41], or to use a drug combination [21, 42].
The BM microenvironment contributes significantly to drug resistance in both hematologic malignancies and solid tumors that metastasize to the BM [10–12]. Conditions in the BM niche that contribute to reduced drug sensitivity might include SFs, stromal cells, and extracellular components [10, 13, 14, 43]. Thus, modulation of signaling pathways involved in mediating the interaction, adhesion, or homing of hematopoietic cancer cells to BM stromal cells is expected to influence drug sensitivity in hematopoietic malignancies [13–16].
The BM microenvironment-mediated drug resistance might be due to SFs or to cell adhesion to the microenvironment compartment. To address BM microenvironment-mediated drug resistance, we monitored the ability of CM collected from MSCs to affect the sensitivity of CML cells to imatinib and crizotinib. We monitored the viability of CML cells as well as induction of PARP cleavage (Figures 1, 2), a marker of apoptosis induction [25].
MEG-01 cells were sensitive to imatinib and crizotinib as evidenced by inhibition of CML cell viability (Figures 1 A, B) and induction of PARP cleavage (Figures 2 B, C). However, CM collected from MSCs conferred resistance to imatinib, consistent with previous observations [16]. Interestingly, MEG-01 cells were more sensitive to MS-5 CM than K562 cells. The presence of CM from MSCs reduced the sensitivity of the CML cells to imatinib-induced apoptosis, but not to crizotinib, suggesting that mesenchymal SFs can promote imatinib resistance with continued sensitivity to the activity of crizotinib (Figure 2). We speculated that drug resistance stimulated by the CM is mediated by SFs secreted by the MSCs. We identified a number of SFs that were present in significant amounts in the CM, such as IL-3, IL-1Rα, IL-17 and IL-12 p70, and speculated that one or a combination of these is responsible for mediating imatinib resistance in CML cells. Our speculation was supported by previous reports showing that IL-7 secreted by MSCs in the BM might protect leukemic cells from apoptosis induced by imatinib through the JAK/STAT-signaling pathway [44]. In addition, Jiang et al. [45] demonstrated autocrine production and activity of IL-3 and granulocyte colony-stimulating factor (G-CSF) in CML [45]. Moreover, culturing K562 cells with HS-5-derived CM significantly inhibited apoptosis induced by imatinib via a STAT3-dependent mechanism [18], and imatinib sensitivity was restored by exposure to STAT3 inhibitor [46]. Our results are also corroborated by Zhang et al. [43], who found that CML stem cells demonstrate increased IL-1 receptor expression and an enhanced signaling response; however, inhibition of IL-1 signaling when combined with TKIs enhanced anti-CML efficacy. In our study, we also monitored levels of activated STAT proteins and found that the presence of MSC CM results in activation of STAT3 and STAT6 (Figure 4). Increased activation of STAT3 has been associated with malignant cell transformation and drug-resistant tumors [44, 46, 47]. Moreover, exposure of CML cells to MSC CM caused an increase in pSTAT3 and consequently increased the expression of STAT3-regulated genes such as Bcl-xl, Mcl-1, and survivin [46]. Thus, we argue that exposure to SFs that stimulate activation of STAT3 or STAT6 promotes the expression of a variety of antiapoptotic genes [48, 49], and hence promotes drug resistance.
JAK family kinases are the upstream activators of STAT proteins and also participate in mediating the function of a variety of cytokines [50]. Thus, we monitored the ability of the JAK1/2 inhibitor ruxolitinib [37] to abolish the drug resistance mediated by MSC CM. The presence of ruxolitinib restored partial imatinib sensitivity in CML cells exposed to MS-5 CM. Our results are consistent with data reported by Mencalha et al. [51] showing that STAT3 inhibitor has an additive effect with imatinib in inducing apoptosis in CML cells, suggesting a potential therapeutic value to combining these two drug regimens for the treatment of CML patients. Moreover, evidence for the potential of drug combinations in CML therapy was also provided by Ma et al. [52], who showed inhibition of growth and proliferation, cell-cycle blockade, and induction of apoptosis in K562 cells transduced with STAT3 siRNA. Thus, the use of two components was recommended to overcome SF-mediated drug resistance: one to inhibit BCR/ABL and the other to inhibit JAK1/2 activity. However, with the approval of multitarget kinase inhibitors, one might select those that are capable of inhibiting both relevant targets. Previously, we demonstrated that crizotinib efficiently inhibits kinase activity of native and T315I-mutated BCR/ABL [29]. In this study, we also found that crizotinib inhibits JAK2 activity. However, crizotinib activity against JAK1 remains to be determined. Crizotinib’s ability to inhibit JAK2 enables it to overcome JAK2-dependent SF-mediated drug resistance. Similarly, ponatinib as a multi-kinase inhibitor was active in overcoming SF-mediated drug resistance in MEG-01, in part by targeting JAK2 activity, probably indirectly (Figure 5). The exact mechanism by which ponatinib inhibited clonogenicity of Ba/F3 JAK2 V617F remains to be elucidated.
In conclusion, the study showed that SFs secreted from MSC, such IL-3 and IL-7, among others, are capable of activating the JAK/STAT pathway and consequently compromise the apoptosis-inducing activity of imatinib targeting CML cells. On the other hand, the multi-kinase inhibitor crizotinib, an FDA-approved drug for ALK-positive non-small cell lung carcinoma targeting ALK kinase, was found to inhibit BCR/ABL kinase activity and is also active in inhibiting JAK2. Exposure to crizotinib actively overcame drug resistance in CML mediated by SFs secreted from MSC. Our results raise the possibility of therapeutic use of crizotinib for CML patients who also require a JAK2 inhibitor to overcome BM microenvironment-mediated drug resistance.