Introduction
An antigen (a) associated with low-density lipoprotein (LDL), called lipoprotein(a) [Lp(a)], was discovered by Kåre Berg in 1963 [1]. Lp(a) is a specific lipoprotein that contains apolipoprotein(a) [apo(a)]. Apo(a) is a polymorphic glycoprotein with carbohydrate-rich moiety which mRNA is expressed almost entirely in the liver [2]. The LPA gene encoding apo(a), with a homology up to 80% with the human plasminogen gene, was sequenced in 1987 by the group of Richard M. Lawn [3].
Lp(a) is a well-established genetically determined risk factor for premature atherosclerosis and its sequelae [4–6]. It has been shown that high Lp(a) in circulating blood, usually defined as values above 50 mg/dl (125 nM), is associated with increased risk of coronary artery disease (CAD), myocardial infarction (MI), ischemic stroke, peripheral arterial disease (PAD), calcific aortic valve disease (CAVD), and heart failure [7]. Inconsistent data link Lp(a) to venous thromboembolism (VTE). At long-term follow-up an increased risk of recurrent major adverse cardiac and cerebrovascular events (MACE) persists after the first episode in patients with hyperlipoproteinemia(a) in comparison with the low Lp(a) population [8].
Mechanisms underlying the increased risk of arterial thromboembolism are still unclear and reach beyond processes typical of atherosclerosis as low-grade lipid-driven inflammation. This overview summarizes the current knowledge on clinical and molecular mechanisms linking Lp(a) with thrombosis at various locations along with future perspectives of emerging therapy reducing Lp(a).
Structure
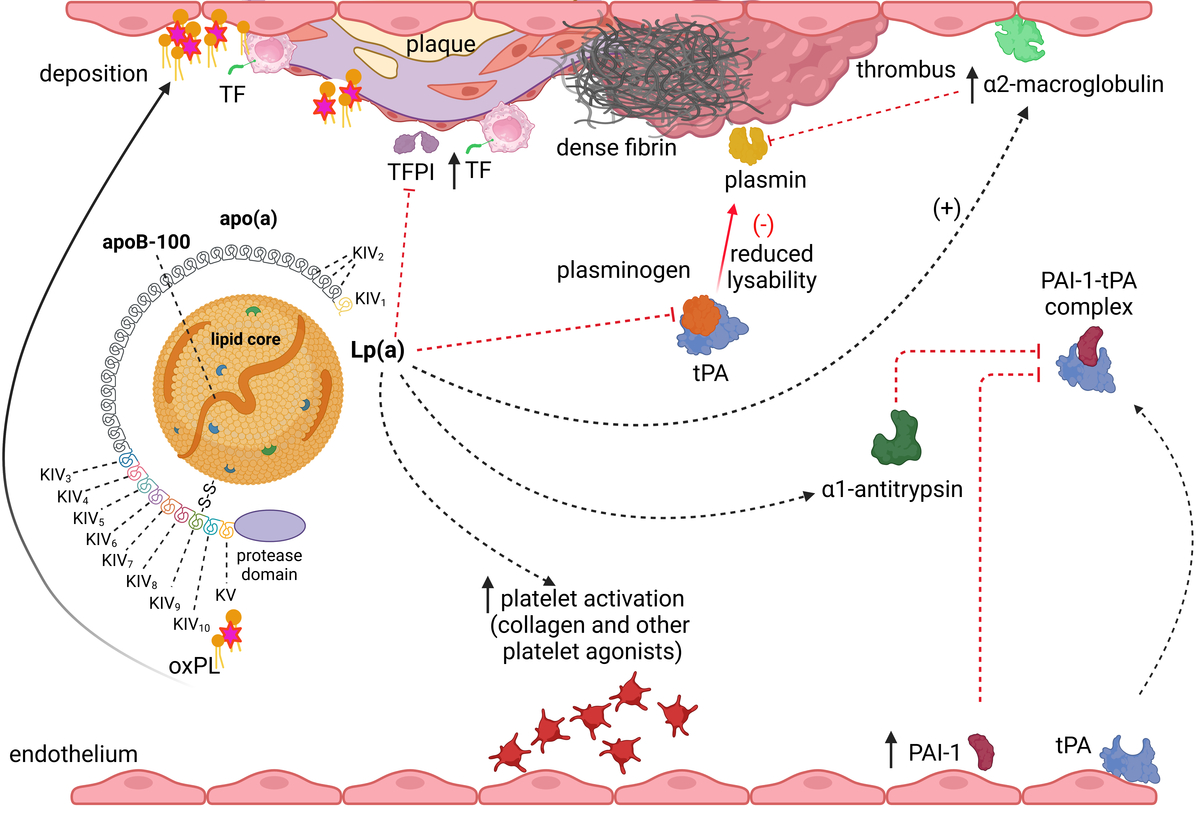
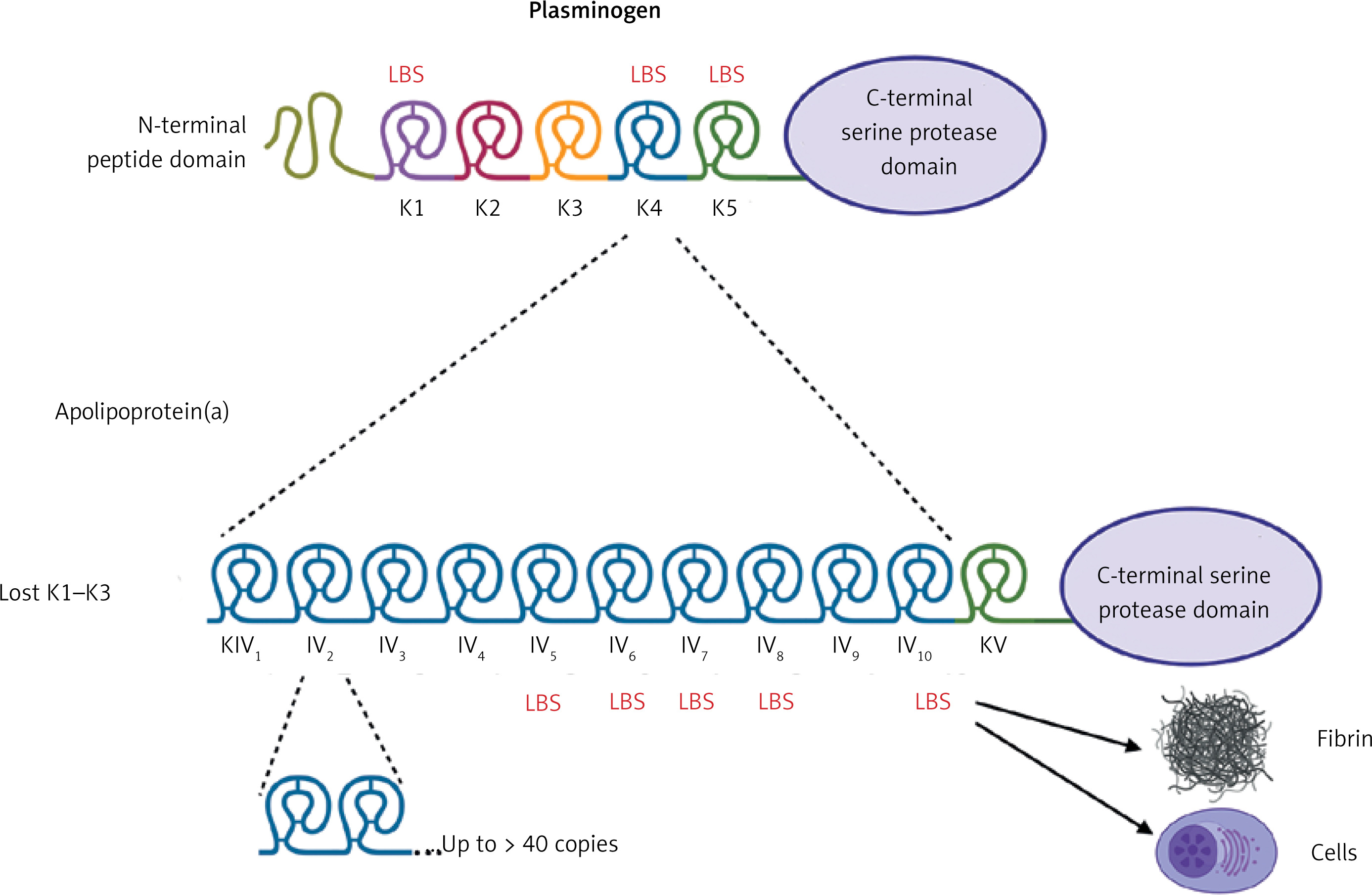
Lp(a) (~3002 kDa) is a particle structurally similar to a regular LDL, containing a core of cholesterol esters and triglycerides surrounded by a monolayer of phospholipids, free cholesterol, and apolipoprotein B-100 (apoB-100) encircled by apo(a) which is bound via a single disulfide bond (Figure 1) [2, 9]. Apo(a) has a unique structure with loop-like structures, termed kringles, including 10 subtypes of KIV (with KIV-1 and KIV-3–10 present in 1 copy, and KIV-2 present in 1 to > 40 copies), 1 copy of KV, and an inactive protease domain (Figure 1) [2, 9]. Unlike apoB, apo(a) does not contain lipid domains. Each individual inherits two copies of the LPA gene, one from each parent, therefore up to 80% of individuals carry two alleles being heterozygous for apo(a) [2]. The KIV type 2 (KIV-2) is encoded in a variable number of tandemly repeated copies in LPA, resulting in different apo(a) isoforms in humans (Figure 1) [2, 9]. Due to the fact that smaller apo(a) forms are easily secreted by the liver, the lower number of KIV-2 is associated with higher Lp(a) levels [2, 9]. Apo(a) isoforms display a structural and functional homology with plasminogen with KIV as the most homologous domain (Figure 1) [2, 9].
Figure 1
A homology between plasminogen and apolipoprotein(a). Plasminogen is structured into seven domains, including the N-terminal peptide domain, five kringle domains, and the C-terminal serine protease domain, which is activated by a single cleavage by tissue- or urokinase-type plasminogen activator. In apolipoprotein(a) the protease domain has a lack of activity. The kringle (K)5 domain of plasminogen is retained as a single copy (KV) in apo(a), while K1-K3 are absent. The K4 domain of plasminogen has evolved into 10 subtypes (KIV-1 to KIV-10) in apo(a). Notably, KIV-2 exists in multiple copies. Weak and strong lysine binding sites (LBS) are indicated by thin and bold markings, respectively. Created in BioRender. Hojda, A. (2024) https://BioRender.com/g19b716
Synthesis and clearance of Lp(a)
Lp(a) synthesis and metabolism are primarily regulated by genetic factors, and the levels of Lp(a) seem to be relatively stable throughout life. It has been recently reported that there is 25% variability in Lp(a) in relation to diet, hormone replacement therapy or inflammation, especially at 30–70 mg/dl levels [9, 10]. The Lp(a) synthesis takes place in the liver in a two-step process – the largely genetically dependent protein synthesis with post-translational modification in the endoplasmic reticulum followed by glycosylation in the Golgi apparatus [11]. The synthesis of Lp(a) proteins and consequently its concentration is related to the coded sequences with numerous different variants modified by single nucleotide polymorphisms and kringle IV type 2-like repeats generating over 40 isoforms of various size [12]. However, other loci (APOH, CETP) can have an effect on Lp(a) expression and concentration in post-translational processing [11]. The LPA gene (6q26) includes a genetic variant (rs3798220) that leads to an Ile to Met substitution in the protease-like domain of apo(a). The Met allele has been linked to higher plasma Lp(a) levels, smaller apo(a) isoforms, and an increased risk of CAD [12]. Notably, heterozygous individuals with the Ile4399Met variant showed elevated Lp(a) levels, a higher risk of CAD, and responded better to aspirin therapy compared to non-carriers [13].
Lp(a) has a relatively long half-life, estimated to be 3 to 4 days in humans, which is longer than LDL, contributing to the persistence of elevated Lp(a) levels in individuals with high genetic predisposition [14]. The exact pathways for Lp(a) clearance are not fully understood, but it is believed that Lp(a) is taken up by multiple tissues and organs, including the kidneys and liver [10, 14].
Lp(a) concentrations
It is estimated that up to 20% of people worldwide have levels of Lp(a) ≥ 50 mg/dl (125 nM) and a very high concentration of Lp(a) > 180 mg/dl occurs in 1% [15, 16]. In Europeans, the prevalence of elevated Lp(a), defined as concentration over 30 mg/dl, reached 26.6% in Germany, 15.9% in Finland, 19.2% in Greece, while 35.0% in the United States [17–20]. In Poland, 17.8% of 800 individuals with a median age of 49 years (interquartile range: 44–53 years) had Lp(a) levels > 50 mg/dl [21]. Blacks have the highest Lp(a) level of all race/ethnic groups studied followed by South Asians, Whites, Hispanics, and East Asians [22]. Women, especially those aged > 50 years, tend to have higher Lp(a) levels compared to men and this difference is more pronounced after menopause [23, 24]. In Caucasians a mean Lp(a) level is about 10 mg/dl [25]. It has been suggested that Lp(a) increases with age, however, such data are scarce and reported for very selected populations [26]. Moreover, atrial fibrillation, hyperlipidemia, and hypertension were independently associated with elevated Lp(a) levels in 2475 primary prevention patients referred to outpatient cardiology units [24]. Given previous studies, variability of serum Lp(a) is > 25% in about 41% of 432 subjects at a median age of 50 years during 1-year follow-up [27]. Given that elevated Lp(a) is an established risk factor for CVD, fluctuations in Lp(a) levels over time can influence both risk assessment and management strategies [28]. This is particularly relevant with ongoing Lp(a)-lowering clinical trials using specific cut-off values for inclusion.
Lp(a) as a prothrombotic factor
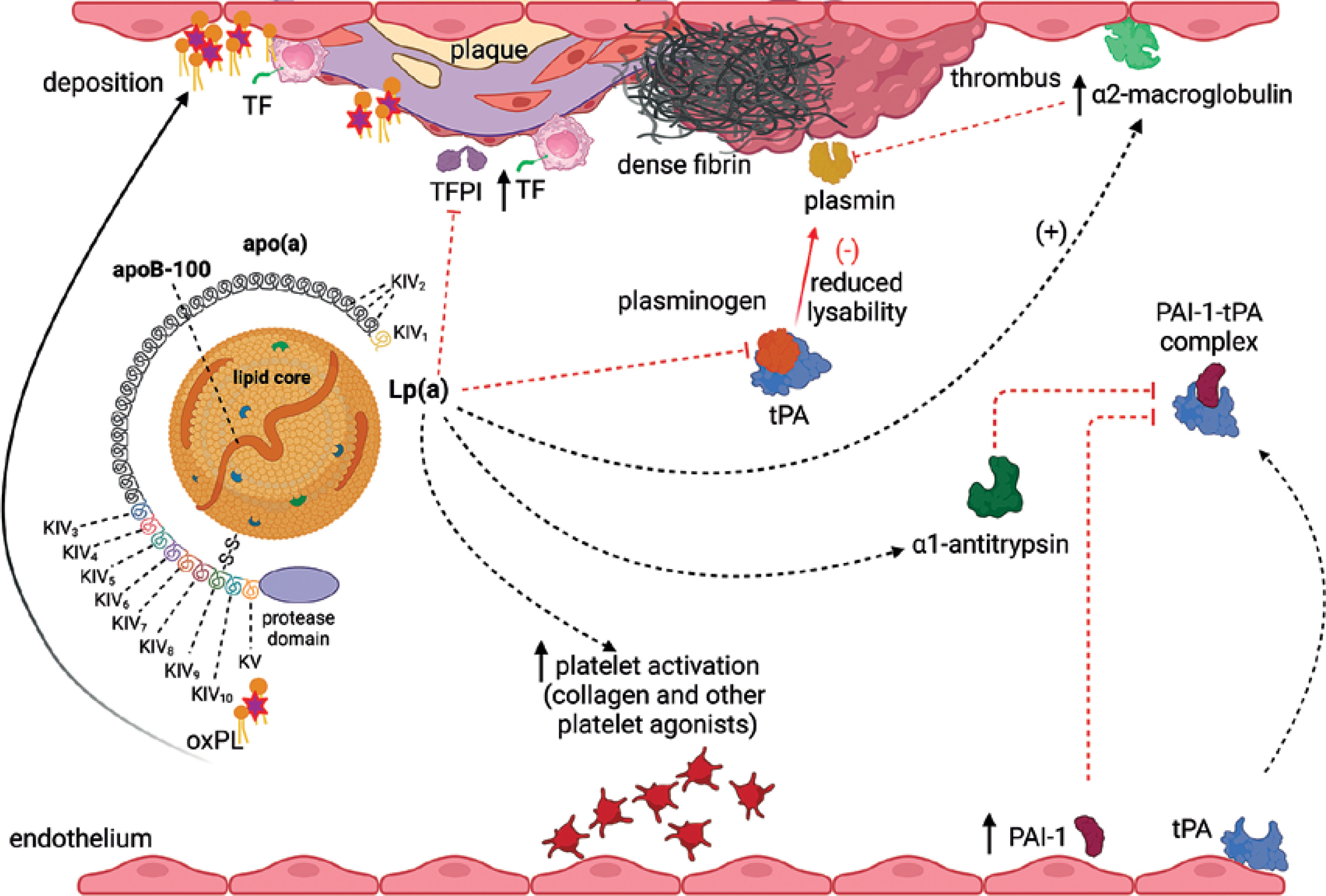
Multiple prothrombotic mechanisms reported in animal and human studies on Lp(a) have been presented in Figure 2.
Figure 2
Prothrombotic actions of Lp(a). Increased circulating Lp(a) levels associate with enhanced platelet activation and tissue factor (TF) expression by macrophages, though evidence is limited. Moreover Lp(a) inhibits tissue factor pathway inhibitor (TFPI), which enhances procoagulant effects of TF. Moreover/Furthermore, Lp(a) at high concentrations can bind to fibrin resulting in formation of denser fibrin structure, resistant to fibrinolysis. Plasminogen is activated within a ternary complex consisting of fibrin and tissue plasminogen activator (tPA), leading to the formation of plasmin, which promotes thrombolysis. Plasminogen activation is hindered through several mechanisms: Lp(a) competes with plasminogen and tPA for binding to fibrin. Additionally, Lp(a) upregulates plasminogen activator inhibitor type 1 (PAI-1) expression within endothelial cells and associates with increased α1-antitrypsin synthesis, both of which inhibit tPA. Furthermore, Lp(a) can bind to α2-macroglobulin, a known non-specific plasmin inhibitor. Created in BioRender. Hojda, A. (2024) https://BioRender.com/b77m703
Impaired fibrinolysis and increased PAI-1 expression
Antifibrinolytic properties of Lp(a) are complex and unclear despite more than 30 years of extensive research. The first concept was based on 80% homology of apo(a) to human plasminogen (Figure 1) leading to the observation that Lp(a) can interfere with plasmin formation [29]. Plasminogen is activated by forming a ternary complex with tissue plasminogen activator (tPA) and fibrin, after which plasmin is released to cleave fibrin. However, in the presence of Lp(a), the apo(a) fragment binds to fibrin, creating a quaternary complex that significantly slows down the rate of plasminogen activation. A biological role of kringles in the Lp(a) molecule is linked to ligand interactions, mostly with lysine-containing substrates [2, 14]. Lysine binding sites (LBS) in KI have the highest affinity for lysine analogs, followed by KIV and KV [30]. Apo(a) binding to fibrin relies on the KV region and the strong LBS in KIV type 10 but does not involve the inactive plasminogen-like protease domain [30]. Small apo(a) isoforms exhibit greater affinity for fibrin than larger isoforms. Lp(a) interacts with fibrinogen, fibrin, and fibrin degradation products, since lysine residues provide binding sites for apo(a) [14]. All the three alpha, beta, and gamma of fibrinogen have been found to interact with Lp(a) [31]. Lp(a) also competes with plasminogen and tPA for binding fibrin, thereby preventing plasmin-mediated clot breakdown and promoting a prothrombotic state [32]. In addition to disrupting ternary complex formation, Lp(a) can influence plasminogen activation by interacting with inhibitors of its components. Lp(a) has been shown to reduce tPA secretion from endothelial cells [33]. Regardless of its impact on tPA secretion, Lp(a) has been reported to increase plasminogen activator inhibitor-1 (PAI-1) expression in HUVEC and human coronary artery endothelial cells via a protein kinase C (PKC)-dependent pathway [34], with effects that can be enhanced by oxidized or glycated Lp(a) [35]. Additionally, Lp(a) binds to α2-macroglobulin (a plasmin inhibitor), histidine-rich glycoprotein (HRG), and α1-antitrypsin (a tPA inhibitor) [31]. Thus, Lp(a) can impair fibrinolysis through several mechanisms: disrupting the interaction of plasminogen, fibrin, and tPA, reducing tPA availability or upregulating the tPA inhibitor, PAI-1. It is important to note that, on a molar basis, plasminogen is nearly always present in higher amounts than apo(a), which markedly minimizes in vivo potency of this protein in inhibiting plasminogen activity [7]. This excess of plasminogen is essential when considering the role of Lp(a) in CVD, where apo(a) may contribute to thrombosis. A study performed on plasminogen knockout mice overexpressing apo(a) showed high incidence of thrombosis, suggesting that Lp(a) may contribute to thrombotic events independently of plasminogen [36]. In summary, although key mechanistic issues and the impact of Lp(a) isoform size are still unclear, the current literature consistently shows that Lp(a)/apo(a) can hinder fibrinolysis and the activation of plasminogen both within fibrin clots and on endothelial cells.
Unfavorably altered fibrin clot structure
Unfavorably altered fibrin clot properties, the so-called prothrombotic fibrin clot phenotype, involve formation of denser fibrin network with thinner, more branched fibrin fibers and smaller pores, which makes clot relatively resistant to fibrinolysis [37]. Such clot phenotype has been reported in patients with arterial and venous thrombosis [37]. It has been suggested that Lp(a) can unfavorably modulate plasma fibrin clot properties in patients [38, 39], even without a prolongation of clot lysis time [40]. This modulation is at least in part genetically determined. It has been demonstrated that white individuals with the LPA Ile4399Met variant (5.33% of carriers) had higher Lp(a) levels (18.5 [2.8–29.7] vs. 5.5 [3.1–12.9] mg/dl) and formed denser fibrin clots (a decrease of clot porosity by 0.44, 95% confidence interval [CI] 0.26–0.62 per each natural log increase in Lp(a)) that were less susceptible to lysis (a prolongation of clot lysis time by 0.48, 95% CI: 0.30–0.66 min per each natural log increase in Lp(a)) compared to non-carriers [41]. It appears that Lp(a) levels ≥ 30 mg/dl discriminates subjects with unfavorable fibrin features from those with a “healthy clot phenotype”. Such Lp(a) concentrations have been associated with about 20% lower fibrin clot porosity and about 15% prolonged clot lysis time both in healthy individuals and patients following MI [42]. In patients with severe aortic stenosis (AS) at a median age of 67 (59–72) years, Lp(a) at a median level of 11.6 (4.0–60.0) mg/dl explained about 20% of variability in plasma clot lysis time among other lipid profile parameters, however, Lp(a) in the multivariable analysis showed a weaker association with clot lysis time than PAI-1 and oxidized LDL [39]. Available data show that in humans Lp(a) affects the fibrin clot phenotype at Lp(a) levels above 30–50 mg/dl.
Increased platelet activation
Lp(a) has been shown to enhance platelet activation and aggregation, but evidence for such Lp(a) mediated effects is limited and their mechanisms are elusive [43–45]. It has been demonstrated that Lp(a) and apo(a) alone can promote platelet activation via thrombin-receptor-activated hexapeptide (TRAP) acting through protease-activated receptor [43]. Lp(a) has inconsistent effects on aggregation. It has been shown that Lp(a) increases platelet aggregation mediated by arachidonic acid (AA) and TRAP, but not by collagen [44]. Lp(a) levels ≥ 30 mg/dl in 6601 patients after percutaneous coronary intervention were associated with greater clot strength assessed by thromboelastography and enhanced platelet aggregation induced by AA and adenosine diphosphate compared to Lp(a) levels < 30 mg/dl, despite dual antiplatelet therapy with aspirin and clopidogrel [45]. On the other hand, Lp(a), isolated from human EDTA plasma samples of apparently healthy donors, in vitro at concentrations of 20–80 µg/ml inhibited in a dose-dependent manner platelet aggregation induced by platelet activating factor (PAF), potentially due to its interaction with PAF-acetylhydrolase (PAF-AH, currently known as lipoprotein-associated phospholipase A2; Lp-PLA2) [46]. A similar effect of Lp(a) at concentrations of 1–100 mg/dl on collagen-induced platelet aggregation has been observed, depending on the increasing levels of cyclic adenosine monophosphate (cAMP) [47]. Moreover, Lp(a) inhibited fibrinogen binding to platelets in response to PAF [46]. However, more recent data showed that Lp(a) enhanced platelet aggregation independently of Lp-PLA2 [48]. Moreover, Salsoso et al. [48] have reported no effect of Lp(a) on platelet reactivity in CAD patients, even at a concentration ≥ 50 mg/dl. These contradictory effects may depend on different platelet agonists and the interactions between Lp(a) subunits (apo(a) and apoB-100/LDL). An impact of Lp(a) on platelets can be however modified by posttranslational modifications, such as lipid peroxidation or acetylation, highlighting the complexity of Lp(a)-platelet interactions [49].
Oxidative stress and inflammation
Oxidative stress generates reactive oxygen species (ROS), which can damage endothelial cells and promote a pro-thrombotic environment. Moreover, inflammation triggers the release of cytokines and other inflammatory mediators, further disrupting vascular integrity and activating platelets and blood coagulation. Lp(a) is considered a pro-inflammatory lipoprotein largely via enhanced oxidative stress. The expression of Lp(a) is upregulated by the proinflammatory cytokine interleukin-6 (IL-6), which binds to multiple sites in the apo(a) promoter in the LPA gene, suggesting that Lp(a) might function as an acute-phase reactant [50]. Lp(a) is known to stimulate the secretion of inflammatory cytokines, attract inflammatory cells, facilitate transendothelial migration, and probably most important, to carry oxidized phospholipids (OxPL). OxPL are typically associated with apoB-100, but within the Lp(a) molecule, their binding depends on the KV of the apo(a) component [51]. The amount of OxPL bound to apo(a) remains constant regardless of apo(a) size, indicating that this binding occurs during synthesis in the liver and not from plasma LDL containing apoB-100 [51]. While this binding may – at low concentrations – play a potential anti-inflammatory role, in individuals with high Lp(a) levels, it could lead to the deposition of OxPL in the vessel walls, contributing to local thrombogenesis [51]. OxPL deposition results from competing action between apo(a) and Lp-PLA2 for OxPL binding, favoring the former reaction. Absence of apo(a) reduced this effect, underlying a dose-dependent manner of Lp(a) action [52]. Moreover, OxPL as well as oxidized Lp(a) can damage the endothelium and activate monocytes and macrophages, which upon activation with inflammatory cytokines (e.g., IL-6, tumor necrosis factor-α, and IL-1β) can express tissue factor (TF) on their surface.
TF-mediated coagulation
TF is a key initiator of blood coagulation in vivo, leading finally to thrombin generation and fibrin formation. An in vitro study showed that monocytes isolated from peripheral blood of healthy individuals treated with Lp(a) or recombinant apo(a) (each 100 nM, encountering concentrations > 30 mg/dl in vivo) doubled TF expression resulting from macrophage-1 antigen (Mac-1; integrin αMβ2) and the NF-κB signaling pathway activation [53]. In contrast to normal arteries, all types of atherosclerotic lesions (from early to advanced) were rich in apo(a) colocalizing in 97% with Mac-1 expression, regardless of age, gender, atherosclerotic risk factors or comorbidities [31]. A similar effect of Lp(a) was not observed in endothelial cells [54]. An actual impact of this mechanism on the risk of thrombosis is unclear.
Reduction in TFPI
Besides enhancing TF expression in monocytes, Lp(a) may also inhibit tissue factor pathway inhibitor (TFPI). In 2001, Caplice et al. [54] showed that nanomolar levels of Lp(a) inhibit TFPI via the apo(a) component, and both TFPI and apo(a) have been found colocalized in smooth muscle cell-rich regions of atherosclerotic plaques, indicating potential interactions. Of note, 90% of both free and cell-bound TFPI in plasma is bound to lipoproteins, including Lp(a), which therefore exert procoagulant effects [55]. In a study performed in 136 CAD patients, serum Lp(a) levels correlated positively with total TFPI concentrations (r = 0.28, p < 0.05) [56]. Moreover, Di Nisio et al. [56] showed about 15% higher TFPI antigen in CAD patients with Lp(a) levels > 30 mg/dl compared to those with Lp(a) < 30 mg/dl.
Lp(a) in arterial thromboembolism
Coronary artery disease
Mendelian randomization studies and genome-wide association studies have conclusively shown that elevated Lp(a) levels are associated with a higher risk of CVD [57]. The association between Lp(a) levels and several cardiovascular outcomes have been reported in recent years (Table I). Moreover, both in the secondary and primary prevention setting among middle-aged UK Biobank participants, elevated Lp(a) levels were associated with an increased risk of MACE [58].
Table I
Clinical studies describing the association of Lp(a) with arterial and venous thromboembolism.
| Author, year | Study design | Population | N | Findings | Ref. |
|---|---|---|---|---|---|
| Arterial thromboembolism | |||||
| Patel, 2021 | Cohort prospective | UK Biobank Participants aged 40–69 years when recruited in 2006–2010 | 460 506 | Lp(a) predicted ASCVD incident among middle-aged adults within primary and secondary prevention contexts HR = 1.11, 95% CI: 1.10–1.12 per 20 mg/dl (50 nmol/l) Lp(a) increment | [58] |
| Kamstrup, 2008 | Cohort prospective | The Danish Copenhagen City Heart Study Individuals aged 20 to 100 years recruited using the Danish Civil Registration System | 9 330 | Lp(a) levels > 120 mg/dl (300 nmol/l) (> 95th percentile) predicted a 3- to 4-fold increase in the risk of MI and absolute 10-year risks of 20% and 35% in high-risk women and men | [59] |
| Dyrbuś, 2024 | Registry | Patients with a very high CV risk | 2 001 | The prevalence of increased levels of Lp(a) > 30 mg/dl (75 nmol/l) were the highest in patients with a history of previous MI (20.6% vs. 14.9%; p = 0.002), with chronic CAD (52.2% vs. 41.5%; p < 0.001) and in patients undergoing PCI during hospitalization (23.9% vs. 19%; p = 0.01) | [60] |
| Rallidis, 2018 | Case-control | Patients with a history of ACS | 1 457 | Elevated Lp(a) > 50 mg/dl (125 nmol/l) was associated with a 3-fold higher likelihood of ACS in younger adults (< 45 years) and a 2-fold higher likelihood in middle-aged (45–60 years) adults, but there was no association in the older participants (> 60 years) | [64] |
| Erqou, 2009 | Meta-analysis of 36 prospective studies | Individuals with no history of CAD and stroke | 126 634 | Each 3.5-fold Lp(a) level increase was associated with a 10% increased risk for ischemic stroke and a 6% increased risk for hemorrhagic stroke | [68] |
| Langsted, 2019 | Cohort prospective | The Danish Copenhagen City Heart Study and General Population Study | 60 512 | HR for stroke was 1.60, 95% CI: 1.24–2.05 for individuals with Lp(a) > 93 mg/dl (> 199 nmol/l) | [70] |
| Arnold, 2021 | Cohort prospective | Patients with AIS | 1 733 | Lp(a) ≥ 40 mg/dl (100 nmol/l) was associated with an increased risk of recurrent events among patients who were either aged < 60 years (adjusted HR = 2.40, 95% CI: 1.05–5.47), had evident LAA stroke etiology (adjusted HR = 2.18, 95% CI: 1.08–4.40), or had no known atrial fibrillation (adjusted HR = 1.60, 95% CI: 1.03–2.48) | [76] |
| Venous thromboembolism | |||||
| Dentali, 2017 | Systematic review and meta-analysis (14 studies) | Adult patients with DVT and/or PE | 2 824 | Lp(a) was slightly but significantly associated with an increased risk of VTE (OR = 1.56, 95% CI: 1.36–1.79; 10 studies) | [92] |
| Wang, 2015 | Cohort prospective | Patients with first-ever SCI | 279 | Elevated Lp(a) ≥ 30 mg/dl incidents were independent predictors of DVT, (OR = 10.35, 95% CI: 2.37–45.35) | [93] |
| Kamstrup, 2012 | Cohort prospective | The Danish Copenhagen City Heart Study and General Population Study | 38 753 | No association of Lp (a) levels nor LPA KIV2 repeats with the risk of VTE | [94] |
| Danik, 2013 | RCT, 100 mg of aspirin or placebo and then monitored them for 10 years for a first MACE | Initially healthy women 45 years of age or older | 21483 | Only extremely high Lp(a) (≥ 99th percentile or 130.6 mg/dl) levels were significantly associated with provoked VTE (HR = 2.55, 95% CI: 1.13–5.7). None of the LPA genetic variants were associated with VTE | [95] |
| Larsson, 2020 | Cohort prospective | UK Biobank participants | 367 586 | No relationship between genetically elevated Lp(a) and DVT | [96] |
| Skuza, 2019 | Cohort prospective | Patients with the first CVST after anticoagulation withdrawal | 80 | Patients with Lp(a) > 30 mg/dl (75 nmol/l) had 3.9-fold higher risk of recurrent CVST | [102] |
| Kuhli-Hattenbach, 2017 | Cohort retrospective | Patients with RVO | 100 | Lp(a) levels > 30 mg/dl (75 nmol/l) were more prevalent among RVO patients (OR = 4.76, 95% CI: 2.0–11.5) | [103] |
| Paciullo, 2021 | Systematic review and meta-analysis | Patients with RVO | 1 040 | Lp(a) levels above normal limits were associated with RVO (OR = 2.38, 95% CI: 1.7–3.34) | [104] |
| Russell, 2023 | Retrospective | TriNetX US Collaborative Network | 35 687 | Lp(a) > 30 mg/dl (75 nmol/l): no association with RVO was found | [105] |
[i] ASCVD – atherosclerotic cardiovascular disease, HR – hazard ratio, CI – confidence interval, Lp (a) – lipoprotein a, MI – myocardial infarction, CV – cardiovascular, CAD – coronary artery disease, PCI – percutaneous coronary intervention, ACS – acute coronary syndrome, AIS – acute ischemic stroke, LAA – large artery atherosclerosis, DVT – deep vein thrombosis, PE – pulmonary embolism, VTE – venous thromboembolism, OR – odds ratio, SCI – spinal cord injuries, RCT –randomized controlled trial, MACE – major adverse cardiovascular events, CVST– cerebral venous sinus thrombosis, RVO – retinal vein occlusion.
The Danish Copenhagen City Heart Study showed a stepwise increase in risk of MI with increasing levels of Lp(a) in both genders. It has been found that high Lp(a) levels > 120 mg/dl (> 95th percentile) predicted a 3- to 4-fold increase in risk of MI and absolute 10-year risks of 20% and 35% in high-risk women and men [59]. A recent report from the Zabrze-Lipoprotein (a) Registry in Silesia, Poland, revealed that the prevalence of increased Lp(a) > 30 mg/dl (75 nmol/l) were the highest in patients with previous MI (20.6% vs. 14.9%; p = 0.002), with chronic CAD (52.2% vs. 41.5%; p < 0.001) and in those undergoing PCI during hospitalization (23.9% vs. 19%; p = 0.01) [60].
The INTERHEART Lp(a) study demonstrated wide interethnic differences in Lp(a) concentration and isoform size. Higher Lp(a) concentrations were associated with an increased risk of MI and carried an especially high population burden in South Asians and Latin Americans. Isoform size was inversely associated with Lp(a) concentration, but in this study did not significantly contribute to risk [61].
Atherosclerotic plaques in patients with elevated Lp(a) levels have a complex morphology, these plaques are prone to repeated ruptures and healing, which can lead to severe and rapid progression of atherosclerosis [62]. Coronary atherosclerosis in patients with high Lp(a) levels mostly manifests clinically as an acute MI, rather than stable angina [63].
Rallidis et al. [64] reported that elevated Lp(a) above 50 mg/dl (125 nmol/l) was associated with a 3-fold likelihood of acute coronary syndrome (ACS) in adults aged < 45 years and a 2-fold likelihood in middle-aged (45–60 years) adults, but there was no association in the older participants (> 60 years). A recent analysis has shown that aspirin use reduced coronary heart disease events (hazard ratio [HR] = 0.54, 95% CI: 0.32–0.94) without significant risk of bleeding in individuals with Lp(a) > 50 mg/dl without CVD at enrollment [65]. Trials on novel agents that substantially reduce Lp(a) are currently underway and will evaluate whether Lp(a) lowering does reduce incidence of MACEs [66].
Stroke
Stroke is the second leading cause of death and a major cause of disability worldwide. Almost 80% of strokes are ischemic strokes (IS) and 15–20% are hemorrhagic strokes (HS) in origin [67]. Numerous data published in recent years have shown that increased Lp(a) levels could be considered as a predictive marker for identifying individuals who are at risk of developing stroke (Table I).
Emerging Risk Factors Collaboration based on 36 prospective studies found that each 3.5-fold increase in the Lp(a) level was associated with a 10% increased risk of IS and a 6% increased risk of HS [68, 69]. The large Danish cohort study showed Lp(a) levels > 93 mg/dl (> 95th percentile) were associated with an increased IS risk compared with Lp(a) levels < 10 mg/dl (HR = 1.60, 95% CI: 1.24–2.05) [70]. Lange et al. [71], who followed patients with acute first-ever IS, demonstrated that after adjustment for potential confounders, patients with elevated Lp(a) levels > 30 mg/dl had a notably higher risk of combined vascular event recurrence (IS, transient ischemic attack [TIA], MI, nonelective coronary revascularization, and cardiovascular death). Meta-analysis of 41 studies performed by Kumar et al. [72] revealed that increased levels of Lp(a) are significantly associated with the risk of IS in adult Asian and Caucasian populations. In the Atherosclerosis Risk in Communities (ARIC) study showed that high Lp(a) was associated with a higher incidence of IS in Blacks and white women, but not in white men [73]. In the REasons for Geographic and Racial Differences in Stroke (REGARDS) study a stronger association between Lp(a) and IS in black than in white participants was reported [74]. Since Lp(a) is an atherothrombotic risk factor, its association with stroke or TIA is likely to be linked to thromboembolic etiology [75]. In the BIOSIGNAL study, elevated Lp(a) was independently associated with large artery atherosclerosis stroke etiology and risk of recurrent cerebrovascular events among individuals aged < 60 years or with evident atherosclerotic disease [76].
Interestingly Lp(a) has been reported to be associated with incident IS in children, which is an uncommon disease (< 1% of children) [76]. Meta-analysis performed by Sultan et al. [77] demonstrated that children with high Lp(a) levels have a 4-fold increased risk of IS with an even higher risk of recurrent event. It is speculated that among the children with elevated Lp(a) the causal mechanism of IS may be disturbed fibrinolysis, as children rarely have atherosclerosis [75].
There are data suggesting that thrombophilic risk factors combined with a higher Lp(a) level amplify the risk of IS and VTE/sinus venous thrombosis [78]. Youyou et al. [79] have suggested that fibrinogen accounts for 10% of the association between Lp(a) and the risk of IS.
Tsimikas [75] has suggested that in young adults with stroke, plaque burden may not fully account for stroke/TIA, and genetically elevated Lp(a) from birth along with superimposed proinflammatory effects of OxPLs and antifibrinolytic properties of apolipoprotein(a) may predispose to ischemic events. Finally, in elderly subjects with stroke, accumulation of multiple risk factors may be additive or synergistic to elevated Lp(a) in manifesting carotid and intracranial disease, leading to distal embolization of plaque/platelet emboli as well as acute thrombotic occlusion [75]. Taken together, elevated Lp(a) increases the risk of IS also without advanced atherosclerosis.
Calcific aortic valve disease
Aortic stenosis (AS) is the most common acquired valvular heart disease in the aging population. The prevalence of severe AS in those aged ≥ 75 years is 3.4% in Europe and the US [80]. Within the stenotic aortic valves, a prominent accumulation of lipoproteins has been observed, being an essential component of CAVD development [81].
In the Danish cohort studies (the Copenhagen City Heart Study and the Copenhagen General Population Study) patients were followed up for 20 years. In patients with Lp(a) levels > 90 mg/dl, the risk of CAVD was 2.9-fold higher than in patients with a low Lp(a) level [82]. The main pathophysiological processes involving Lp(a) in CAVD include endothelial dysfunction, indirect promotion of foam cell formation through OxPL, valvular calcification, as well as inflammation, oxidative stress, and the direct promotion of valvular calcification [83]. OxPLs are co-expressed with Lp(a) within the stenotic leaflets and promote valvular calcification [84].
A genome-wide association study revealed that LPA rs10455872 genetic variant was associated with a 2-fold increased risk of aortic leaflet calcification [83]. Kamstrup et al. [82] demonstrated that OxPL levels were related to the increased risk of AS. Siudut et al. [39] reported that in AS, Lp(a) and oxidized LDL contribute to hypofibrinolysis. Kopytek et al. [85] observed enhanced valvular OxPL expression in patients with increased serum Lp(a) concentrations equal to or above 50 mg/dl. At increased Lp(a) levels OxPLs impaired fibrinolysis by inhibiting tPA-mediated plasminogen activation and inhibition of plasminogen binding to fibrin [86]. There is evidence that hypofibrinolysis is linked to increased AS severity [39, 87], therefore OxPL-lowering therapies could slow down AS progression.
A post-hoc analysis of the FOURIER (Further Cardiovascular Outcomes Research With proprotein convertase subtilisin/kexin type 9 (PCSK9) Inhibition in Subjects With Elevated Risk) trial with evolocumab highlighted the Lp(a) reduction as a therapeutic option for AS. In this analysis, elevated Lp(a) levels were associated with higher rates of AS events including aortic valve replacement.
Patients treated with evolocumab had a numerically lower incidence of new or worsening AS or valve replacement when compared with placebo [88]. Given these findings authors speculate whether targeted Lp(a) reduction may impact AS progression. This is in line with the results from the Rotterdam Study, in which Lp(a) was associated with baseline and new-onset CAVD but not with AS progression, suggesting that Lp(a)-lowering interventions may be most effective in pre-calcific stages of CAVD [89]. Of note, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors were reported to reduce the incidence of cardiovascular events [90].
Venous thromboembolism
VTE is the third most common cause of vascular mortality worldwide and comprises deep-vein thrombosis (DVT) and pulmonary embolism (PE). About two-thirds of VTE episodes manifest as DVT and one-third as PE with or without DVT. Less frequently, thrombosis affects other veins including upper extremity veins, cerebral venous sinuses, and mesenteric, renal, and hepatic veins [91]. Evidence that Lp(a) might represent a risk factor for VTE remains highly controversial (Table I).
Pulmonary embolism and deep vein thrombosis
Meta-analysis performed by Dentali et al. [92] showed a significant but weak association between Lp(a) levels and VTE (OR = 1.56, 95% CI: 1.36–1.79). The study by Wang et al. [93] among individuals with spinal cord injuries demonstrated that increased serum Lp(a) was an independent predictor of DVT in this population. However, in the large Copenhagen City Heart Study and Copenhagen General Population Study, neither tertiles of Lp(a) levels nor LPA KIV2 repeats were associated with incident VTE [94]. Interestingly, post hoc analyses in both populations revealed that extremely high Lp(a) levels (≥ 95th percentile) were associated with VTE occurrence (OR = 1.33, 95% CI: 1.06–1.69) [94]. However, there were no associations between Lp(a) quintile and total, provoked (e.g. by cancer, surgery, or hospitalization) or unprovoked VTE in participants in the Women’s Health Study [95]. Only extremely high Lp(a) levels (≥ 99th percentile or 130.6 mg/dl) were significantly associated with provoked VTE (HR = 2.55, 95% CI: 1.13–5.7). None of the LPA genetic variants were associated with VTE [95]. Similarly, the Mendelian randomization study conducted in the UK Biobank found no significant relationship between elevated Lp(a) and either DVT (OR = 1.00, 95% CI: 0.96–1.04 per 50 mg/dl increase; 9454 cases) or PE (OR = 1.01, 95% CI: 0.95–1.07; 6148 cases) [96, 97].
Meta-analysis of combined data from FOURIER (evolocumab) and ODYSSEY OUTCOMES (Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment with Alirocumab) trials demonstrated a 31% relative risk reduction in VTE with inhibitors and Lp(a) reduction seemed to be an important mediator of this effect [98]. There was an association between baseline Lp(a) concentrations and the magnitude of VTE risk reduction. In patients with higher baseline Lp(a) levels, evolocumab reduced Lp(a) by 13.2 mg/dl (33 nmol/l) and the risk of VTE by 48% (HR = 0.52, 95% CI: 0.30–0.89) whereas, in patients with lower baseline Lp(a) levels, evolocumab reduced Lp(a) by only 7 nmol/l and had no effect on VTE risk. Altogether, association of Lp(a) with VTE appears much weaker and more complex as compared to that for arterial thrombosis.
Venous thromboembolism at atypical sites
Cerebral vein thrombosis (CVST)
In the literature there have been case reports linking CVST to markedly increased Lp(a) levels [99–101] with 1 case of recurrence of CVST [100]. In 2019 Skuza et al. [102] reported that patients with CVST and Lp(a) > 30 mg/dl had a 3.9-fold higher risk of recurrent CVST. It has been found that faster formation of denser plasma fibrin clots displaying reduced susceptibility to lysis characterizes patients with CVST, and importantly, denser clot structure may predispose to recurrence of CVST. It might be speculated that association of elevated Lp(a) with CVST could be at least in part driven by unfavorably altered fibrin clot properties.
Retinal vein occlusion (RVO)
There is some evidence that elevated Lp(a) plasma levels > 30 mg/dl were significantly more prevalent among RVO patients than among controls [103, 104] (Table I).
However, recent data from the TriNetX US Collaborative Network, a large national database showed the lack of association between Lp(a) concentration and the risk of RVO [105].
Portal vein thrombosis (PVT)
Malaguarnera et al. [106] have observed that patients with PVT had higher serum Lp(a) levels than healthy individuals. This small-sized study has not been confirmed by further reports.
Taking into account the prothrombotic and antifibrinolytic properties of Lp(a) and the emerging evidence from both epidemiological and clinical studies; there is a debate that Lp(a) may also be linked to the development of VTE or recurrent VTE, especially in patients with unfavorably altered fibrin clot properties, which have been reported to be associated both with first-ever and recurrent thrombosis [107]. Despite conflicting data one could speculate that the effects of Lp(a) on the VTE risk may be evident only at the highest Lp(a) concentrations [108].
Future perspectives
Current evidence indicates that Lp(a) levels above 30 mg/dl in the US and 50 mg/dl in Europe are associated with an increased risk of MI and stroke, being perceived as an important potentially modifiable contributor among several modifiable and nonmodifiable CV risk factors [109]. Interestingly, Lp(a) has been linked to arterial thrombosis, with some studies suggesting a connection to atypical forms of thrombosis. However, data regarding its role in patients with DVT remain inconsistent, despite several prothrombotic effects of Lp(a) that are common for both venous and arterial thrombosis. Lp(a) containing protein and lipid components, both of which can undergo post-translational modifications in particular oxidation, especially under conditions of heightened oxidative stress could produce prothrombotic effects of varying magnitude depending on environmental factors. It appears that prothrombotic and antifibrinolytic effects of Lp(a) are more pronounced when oxidative stress is significantly elevated linking this lipoprotein with arterial thrombosis largely in atherosclerotic vascular disease. Therefore, this effect may be less relevant in younger patients, in whom oxidative stress has been documented to be counterbalanced by antioxidative mechanisms [110]. Ongoing research explores new drugs, for instance pelacarsen, that can lower Lp(a) levels even by 80% [7, 111], and it can be speculated that they might exert anticoagulant effects and further reduce the risk of thromboembolic events, regardless of the vascular bed involved, like statins do [112]. Advances in genomic, proteomic, and metabolomic research might help better characterize Lp(a) as a factor increasing thrombotic risk and guide novel Lp(a) reducing therapies.