Introduction
Bone is a preferred site for metastasis for numerous cancers, including breast and prostate [1]. Metastasis to bone involves initial transition of epithelial cells to mesenchymal cells and subsequent intravasation to the blood and lymph vessels, and finally hematogenous dissemination to perivascular niches of the bone marrow sinusoids, which are also the site of formation of the metastatic lesions [2]. Protein kinase C epsilon (PKCε) has been shown to potentiate both metastasis to bone and the resultant pain in the bone via the transient receptor potential vanilloid 1 (TRPV1) receptor [2, 3].
The PKC is a family of serine-threonine kinases that have been shown to regulate different aspects of differentiation, proliferation, motility, and apoptosis in cells. This family has ten structurally related isozymes that have been classified as classical or conventional PKCs – α, βI, βII, and γ; novel PKCs – δ, ε, η, and φ; and atypical PKCs – ζ, and λ/ι. Historically even though PKCs have been categorized as oncogenes, recent research has shown that individual isozyme has a context-dependent action as either a pro-oncogenic or an anti-carcinogenic effector [2, 4–6]. However, PKCε has been shown to function as a potential tumor biomarker and oncogenic kinase in all cases, inclusive of breast, lung, prostate, and head and neck cancer in both in vitro and in vivo models [7–14]. Pharmacological PKCε inhibition is thus being touted as a potential therapeutic option in a multitude of cancer metastases [14, 15].
However, the precise mechanisms by which PKCs exert their effect have not been completely defined yet. Hence, the objective of the current study was to investigate how PKCs exert their effect on bone cancer metastasis and to test the effect of pharmacological inhibition of PKC on bone metastasis. Our results reveal that PKC activation actually causes global translational upregulation whereas its inhibition almost completely attenuates bone metastasis in a xenograft model.
Material and methods
Cell culture and treatment
The DAN (CRL-2130) cell line was purchased from the ATCC (Manassas, VA, USA) and cultured in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Shanghai, China) and 100 IU/ml penicillin and 100 μg/ml streptomycin (Thermo Fisher Scientific). The cells were treated with 10 nM Go6983 (Selleckchem, Texas, USA), 10 nM bisindolylmaleimide IX (Bis IX) (Sigma-Aldrich), or the Akti-1/2 inhibitor (Abcam, Waltham, MA, USA) for indicated times to cell harvest and analysis.
Cell lysis and immunoblot analysis
To perform immunoblot analysis, Pierce RIPA lysis buffer (Thermo Fisher Scientific) was used to lyse the cells. The lysis buffer was supplemented with mini protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN, USA) and phosphatase inhibitor cocktails 2 and 3 (Sigma-Aldrich Shanghai Trading Co Ltd, Shanghai, China). Thirty micrograms of protein lysates were resolved by SDS-PAGE. Membranes were probed with indicated primary antibodies at 1 : 1000 dilution and detected by chemiluminescence. Anti-E-cadherin, N-cadherin, P-PKC (Y311), Total PKC, P-Akt (S473), Total Akt, P-S6K1 (T389), Total S6K1, P-S6 (S240/244), and Total S6 antibodies were purchased from Cell Signaling Technology (USA). Anti-fibronectin and β-actin antibodies were purchased from Abcam (Waltham, MA, USA). Anti-puromycin antibody was obtained from Kerafast (Boston, MA, USA).
Immunofluorescence analysis
DAN cells were grown on glass coverslips in 24-well plates overnight prior to treatment with TGF-β (5 ng/ml; Roche Diagnostics) for 72 h. Cells were treated with 1 nM Go6983 or DMSO for the last 24 h. Cells were then washed with PBS and fixed with 4% paraformaldehyde (in PBS) at room temperature (RT) for 15 min or with ice-cold methanol at room temperature (RT) for 7 min. Cells were then washed three times with PBS prior to permeabilization with 0.1% Triton X-100 in PBS for 5 min. Permeabilized cells were washed five times with PBS and blocked with blocking reagent (0.1% saponin, 10% goat serum in PBS) for 1 h at room temperature (RT). Cells were then washed once with PBS, followed by incubation with anti-E-cadherin (1 : 500) and anti-P-SMAD2/3 (1 : 200; Cell Signaling Technology) primary antibodies in blocking reagent overnight at 4°C. Cells were washed three times with PBS and incubated with labeled secondary antibodies for 1 h at RT in the dark. Cells were then washed five times with PBS and coverslips were mounted with Vectashield containing DAPI (H-1200) prior to microscopy. Images were acquired through a 20× objective with a Zeiss 710 confocal laser microscope (Zeiss, Oberkochen, Germany). Representative images are shown in all figures at the same exposure and magnification and in merged color images; co-localization is indicated by yellow and orange regions.
Xenograft assay
Experiments were approved by the Institutional Animal Care and Use Committee of the Chinese General PLA Hospital. For xenograft assays, 106 firefly luciferase expressing DAN cells were injected via the tail vein of six-week-old female p53–/– mice (n = 5 per group). One day after the initial injection, mice were injected with either DMSO or 10 nM/kg body weight of Go6983 (once every day). Mice were assessed weekly for tumor formation up to 28 days using in vivo bioluminescence imaging using an IVIS imaging system (IVIS Imaging System 200, Xenogen Corporation, PerkinElmer, Waltham, USA) fitted with an ultrasensitive CCD camera. After euthanasia of mice on day 28, lungs were excised and imaged by bioluminescence imaging to detect metastatic lesions.
Results
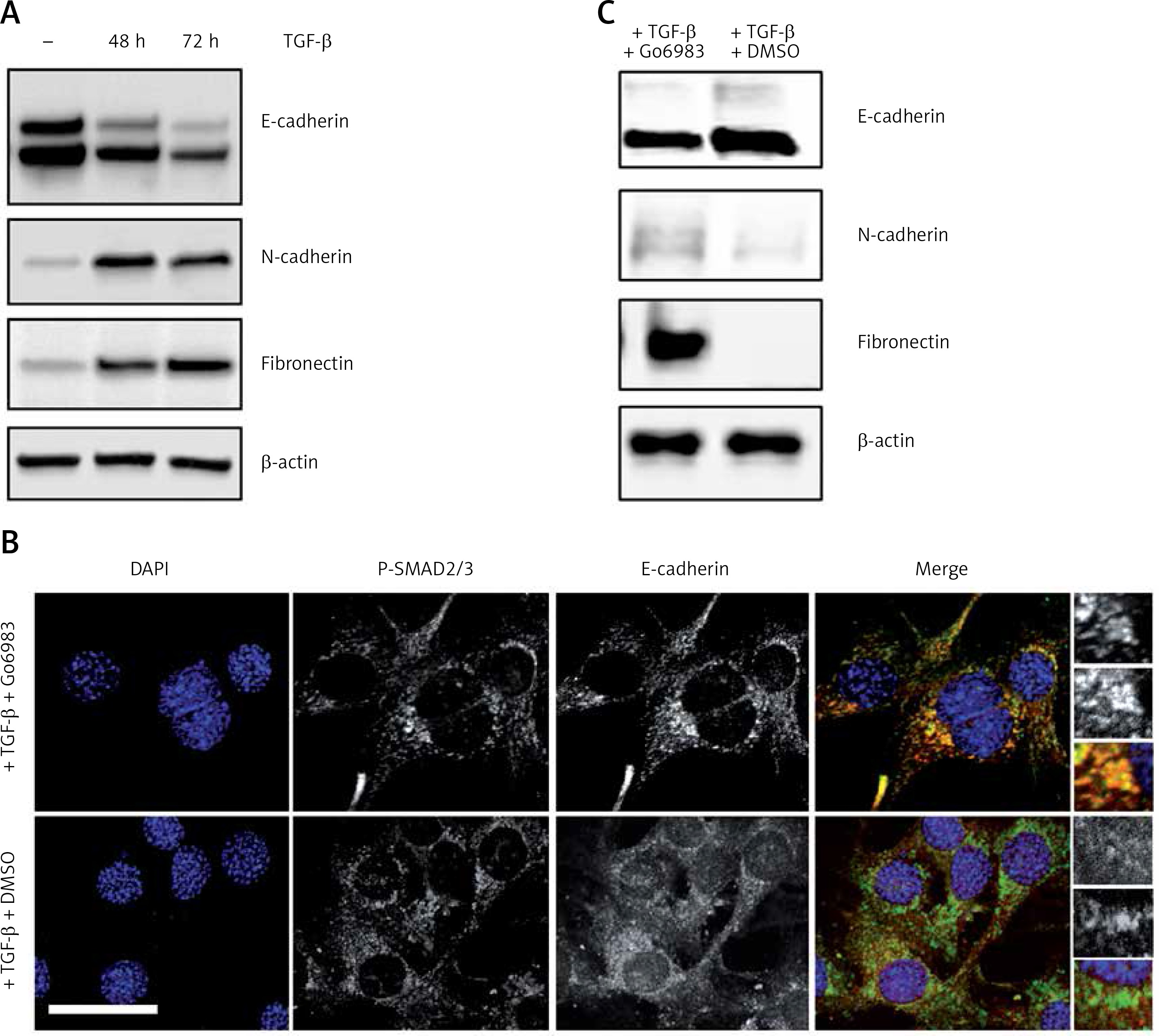
The PKC activity has been shown to be centrally related to bone cancer metastasis [2, 3] and transforming growth factor β (TGF-β) is known to activate PKC [16]. Moreover, a central requirement of metastasis is downregulation of the epithelial cell marker E-cadherin and gain of mesenchymal cell markers such as fibronectin and N-cadherin [17]. Hence, to determine whether treatment with TGF-β of the osteosarcoma cell line DAN would induce changes in epithelial and mesenchymal cell markers, DAN cells were treated with TGF-β over a period of 72 h and cell lysates were probed for E-cadherin, fibronectin, and N-cadherin. Whereas TGF-β resulted in significant loss of E-cadherin expression, it also resulted in concomitant upregulation of fibronectin and N-cadherin (Figure 1 A), indicating that TGF-β treatment does induce a mesenchymal phenotype in these cells. To next determine if this switch between epithelial and mesenchymal phenotype was being mediated by PKC activation, we treated cells with the PKC inhibitor Go6983 [18] for the last 24 h of TGF-β treatment and then performed immunofluorescence for E-cadherin. Treatment with Go6983 prevented E-cadherin expression from being downregulated, even though it did not affect TGF-β activation as evident by P-Smad2/3 staining (Figures 1 B, C). Treatment with Go6983 also inhibited induction of mesenchymal cell markers, N-cadherin and fibronectin (Figure 1 C). However, once cells underwent epithelial-mesenchymal transition (EMT), treatment with the PKC inhibitor Go6983 could not revert the cells to an epithelial phenotype (data not shown). Of note, no significant cell death was observed within 24 h of treatment with the PKC inhibitor at the indicated dose.
Figure 1
Treatment with TGF-β leads to a switch from epithelial to mesenchymal cell marker in the osteosarcoma cell line, which can be inhibited by pharmacological inhibition of PKC. A – Immunoblot analysis of indicated markers in cell lysates obtained from DAN cells treated with TGF-β for 72 h. The blots were stripped and re-probed with anti-β-actin to confirm equal loading. Representative blots from three independent experiments are shown. B – Immunofluorescence analysis of E-cadherin and P-Smad2/3 in DAN cells treated with TGF-β for 72 h and with 10 nM of Go6983 or DMSO in hours 48–72. Scale bar: 10 μm. Images from three independent experiments are shown. C – Immunoblot analysis of indicated markers in cell lysates obtained from DAN cells treated with TGF-β for 72 h and with 10 nM of Go6983 or DMSO from hours 48–72. The blots were stripped and re-probed with anti-β-actin to confirm equal loading
To determine whether TGF-β treatment causes PKC activation in DAN cells, we determined P-PKC (Y311) levels in DAN cells ± TGF-β treatment and saw robust induction of P-PKC (Figure 2 A). This induction was prevented by prior treatment with either Go6983 or another pan-PKC inhibitor, Bis IX (Figure 2 A). Even the endogenous PKC activity seen in DAN cells (Figure 2 B) can be completely inhibited by prior treatment with Go6983 (Figure 2 C).
Figure 2
TGF-β treatment leads to increased PKC-activation which can be effectively attenuated by pharmacological inhibition of PKC. A – Immunoblot analysis of indicated markers in PKC activation (P-PKC, Y311), and Akt activation (P-Akt, S473) in cell lysates obtained from DAN cells treated with TGF-β for 72 h ± PKC inhibitors Go6983 or Bis IX. The blots were stripped and re-probed with anti-β-actin to confirm equal loading. Representative blots from three independent experiments are shown. B – Immunofluorescence analysis of basal PKC activation in DAN cells ± 10 nM of Go6983 for 30 min. Scale bar: 10 μm. Images from three independent experiments are shown
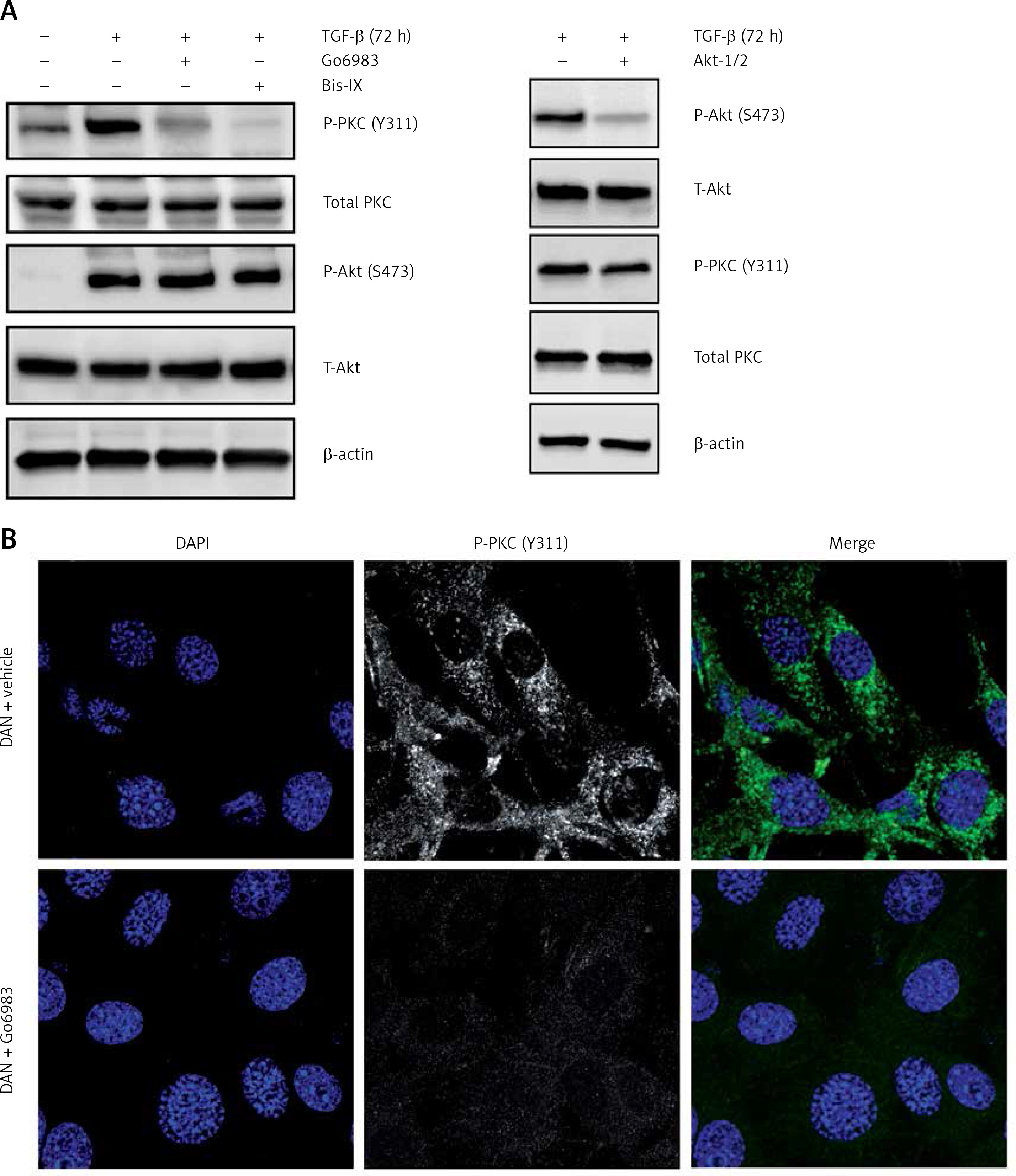
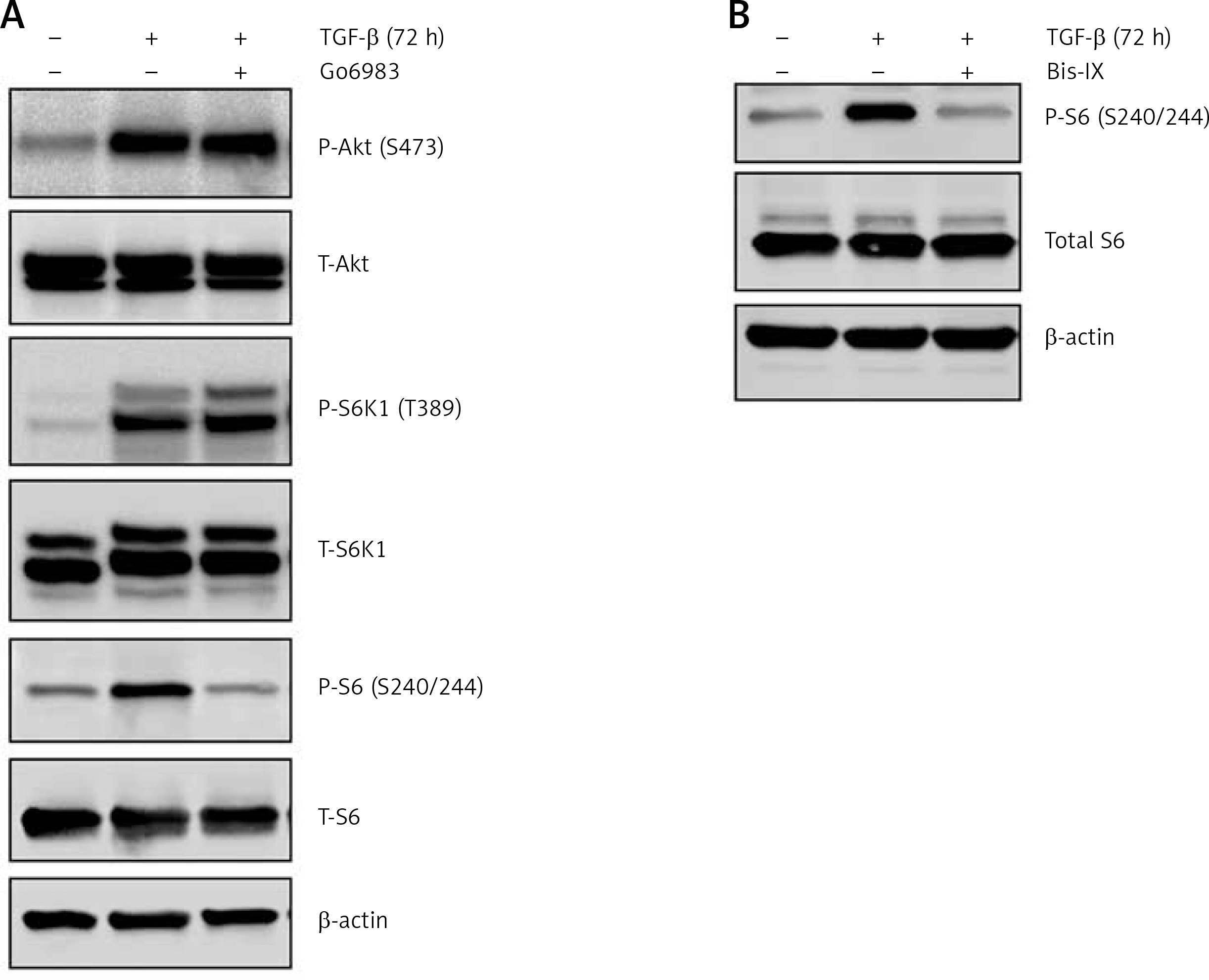
The TGF-β treatment is known to induce Akt activation [19]; however, the effect of Go6983 or Bis IX on PKC activation was independent of changes in Akt activation as assessed by P-Akt (S473) expression (Figure 2 A), indicating that activation of PKC is either downstream of Akt activation or is independent of Akt activation. To determine whether Akt activation occurs upstream of PKC activation, DAN cells were treated with the Akt inhibitor Akti-1/2. Even though Akti-1/2 inhibited Akt activation and EMT, it did not have any effect on P-PKC (Figure 2 D), indicating that Akt and PKC activation occurs by independent pathways following treatment with TGF-β in these cells. Next, we tested whether PKC activation in these cells is reliant on mTORC1 signaling. We found that inhibition of PKC did not decrease the TGF-β-mediated phosphorylation of S6K1; however, TGF-β-mediated activation of ribosomal protein 6 (S6) was abolished by PKC inhibition (Figure 3 A). Similar results were obtained when the other pan-PKC inhibitor Bis IX was used (Figure 3 B). These results are consistent with the previous finding that PKC can phosphorylate S6 independently of mTORC1 activity [20, 21].
Figure 3
TGF-β-treatment-mediated PKC activation results in increased phosphorylation of ribosomal protein S6. Immunoblot analysis of P-S6K1 and P-S6 in cell lysates obtained from DAN cells treated with TGF-β for 72 h ± PKC inhibitors Go6983 (A) or Bis IX (B). The blots were stripped and re-probed with anti-β-actin in each case to confirm equal loading. Representative blots from three independent experiments are shown
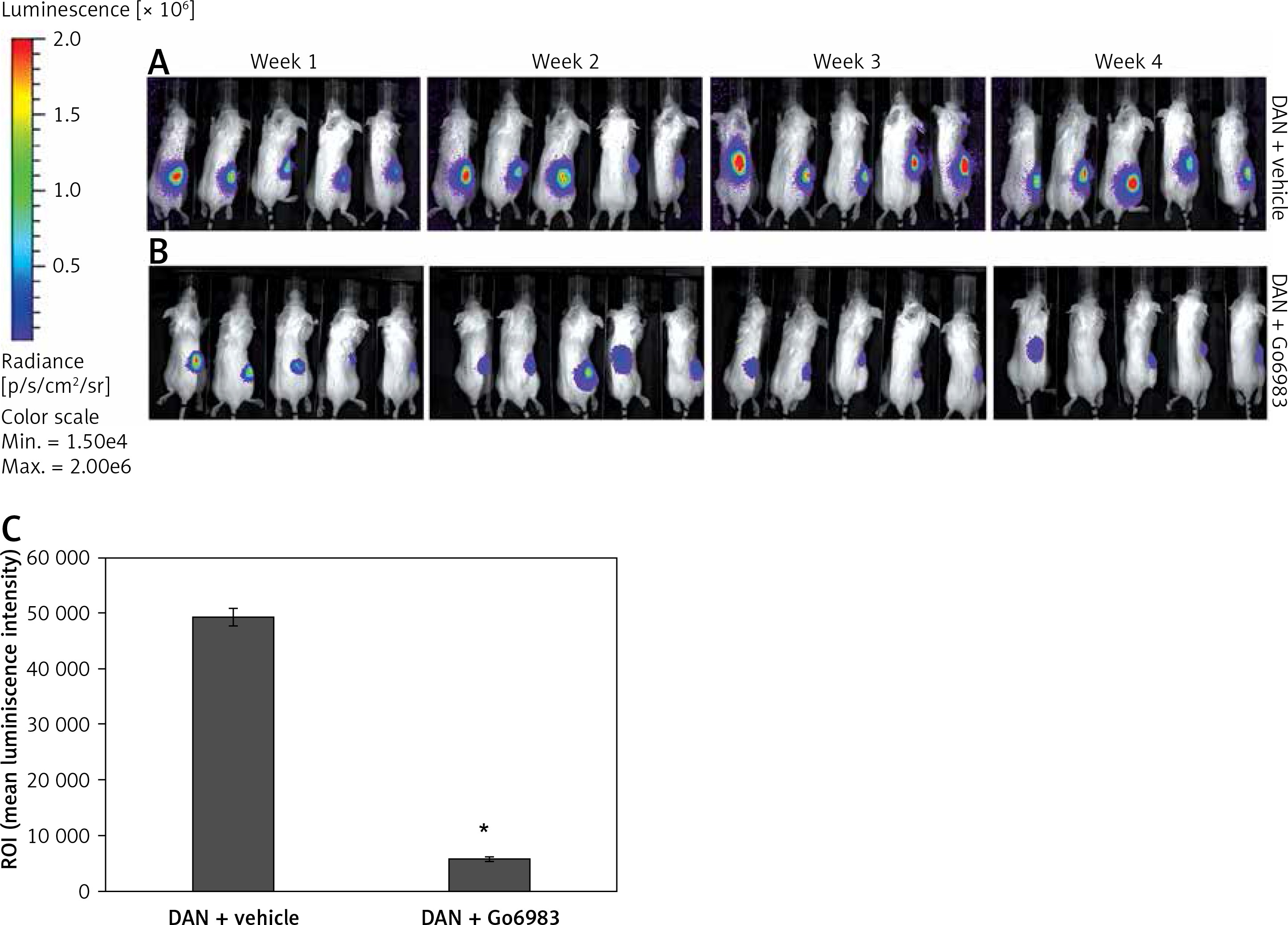
We finally determined whether inhibition of PKC activity can attenuate in vivo metastasis of DAN cells using a xenograft assay. Firefly luciferase labeled DAN cells were injected into the tail vein of p53–/– mice. The mice were injected with either DMSO or Go6983 via the tail vein every day for 28 days and the cells were traced every week for 4 weeks via bioluminescence imaging. Whereas DAN cells formed robust tumors in the hind limbs (Figure 4 A), treatment with Go6983 significantly attenuated these tumors (Figure 4 B). After euthanasia bones were imaged for metastatic lesions, and Go69983 treatment significantly inhibited metastatic lesions in the bones (Figure 4 C). Taken together, our results indicated that inhibition of PKC activity can inhibit tumorigenesis and metastatic progression of bone cancer.
Figure 4
PKC activation correlates with metastatic potential of DAN cells in vivo. A, B – Firefly luciferase expressing DAN cells were intravenously injected subcutaneously via the tail vein in p53–/– mice. From the second day, mice were injected with either DMSO (A) or 1 nM of Go6983 (B). Mice were imaged by in vivo luciferase imaging up to 4 weeks to detect tumor formation. C – After euthanasia, bones from DMSO (vehicle) treated and Go6983 treated mice were excised and imaged by bioluminescence imaging to detect metastatic lesions. The mean luminescence intensity of the region of interest (ROI) is shown
*P < 0.05.
Discussion
Protein kinase C is a prototypical class of serine/threonine kinases that consists of 10 members, which are products of 9 different genes [4]. Extensive work established that PKC is involved in multiple signaling pathways that regulate cell function, including growth, proliferation, differentiation and survival, and dysregulation of PKC activity is often associated with a number of diseases, including cancer and metabolic disorders [4]. Earlier studies have shown PKC regulates eukaryotic translational machinery by activating cellular translation [22–24].
The PKC inhibitors have been sparsely tested in clinical trials partly because of the lack of scientific validation of PKC isoforms being valid targets in cancer given that most studies are correlative at best. Midostaurin was the first PKC inhibitor tested in clinical trials against cancer [25]; however, the results have not been favorable [26]. Enzastaurin had a favorable outcome in phase II trials [27], but was halted mid-way through a phase III trial because of failure to meet the primary end point [28]. So there remains a valid reason for both validating PKC isoforms and their activation as direct targets in different cancers, as well as in finding more potent inhibitors. The role of PKC as a tumor promoter or a suppressor is still not established. While several studies have shown that activation of PKC confers tumor formation, metastasis and invasion, some studies reported that inactive mutations in PKC are associated with disease progression in multiple cancer types [24, 29, 30]. Together, these studies show enormous complexities in understanding the role of PKC in cancer and suggest that the function of PKC as a tumor promoter or a tumor suppressor highly depends on the context of specific oncogenic alterations.
In conclusion, in the current study we saw that PKC activation results in global upregulation of translation in osteosarcoma cells and that potentiates metastasis in vivo. Importantly, treatment with the PKC inhibitor Go6983 results in suppression of P-S6, attenuates global translational upregulation, and reduces the potential of these cells to metastasize in vivo. Even though these are critical findings, the translational potential can actually be realized only when it is determined which isoform of PKC is being affected by Go6983. Moreover, it will be imperative to determine toxicity as well as changes in overall gene expression by RNA sequencing.