Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disease that affects the elderly in most parts of the world. The incidence of PD is increasing in all parts of the world, irrespective of the population [1, 2]. In Europe, it is known to be affecting about 1% of people above 60 years of age [3, 4]. The disease has many motor symptoms such as bradykinesia, rigidity, and postural instability, and non-motor symptoms such as cognitive problems and disturbances in sleep patterns, and total body dysfunction [5]. There is a reduction in dopamine production in the substantia nigra compacta, and its delivery to the dorsal striatum is attributed to the decline in the patients’ movement with PD [6]. This and the accumulation of the alpha-synuclein aggregate protein in the neurons [7] are PD’s principal causes.
Healthy cells have a balanced state of action observed between histone acetyltransferases (HATs) and histone deacetylases (HDACs) in the acetylation and deacetylation of histone proteins and thereby the control of gene transcription [8, 9]. The control of gene expression helps neurons maintain their homeostasis [8]. An imbalance in the activities of HATs and HDACs would result in a pathogenic state in which the balance is tilted towards histone deacetylation, which was observed in another neurodegenerative disease – Alzheimer’s disease. In PD, α-synuclein accumulation promotes histone PD hypoacetylation, as ascertained from both in vitro and in vivo research models [10]. In order to correct the tilt in the balance between HATs and HDACs, HDAC inhibitors are used to reduce cell death in the nigrostriatal pathways in PD [9, 11, 12].
Entinostat is a synthetic benzamide derivative that belongs to the substituted pyridyl carbamate class of HDAC-inhibiting compounds that can be used orally against HDACs I and IV classes [13, 14]. It promotes histone hyperacetylation and helps in the transcriptional activation of specific genes [15] that are involved in cell proliferation, differentiation, and apoptosis. These refer to the HDAC inhibiting activities of entinostat in the control of the solid tumour, and we are interested in testing this synthetic compound in the treatment of PD.
We propose herein that entinostat would act against PD by inhibiting the activity of HDAC and restoring the cellular functions and alleviating the influence of PD biomarkers in the pathogenesis of the disease.
Material and methods
PD rat model
For the study, Wistar male rats (150 ±10 g) were used. The experimental protocol was followed as per the Institutional Animal Ethical Committee’s approval, and all animal experiments were carried out strictly in accordance with the institutional guidelines and suggestions from the Ethics Committee. All animals were kept in cages in a temperature-controlled room with a 12-hour light/dark cycle with temperature and humidity maintained at 25 ±2°C and 55 ±5%, respectively. Rats were allowed free access to regular standard rat chow and water. A total of 48 rats were randomly allocated to 4 groups (n = 12 each): Group 1 as a sham control group; group 2 as a PD group (rotenone dissolved in 1% DMSO at the back of the neck) [14, 15]; group 3 were given entinostat (20 mg/kg) intraperitoneally before the PD induction; and group 4 rats were given entinostat alone as drug control. The rats were maintained for 3 months. At the end of the experimental period, the animals were assessed for behavioural and neurological functions such as an elevated plus maze, forced swim test, Morris water maze, rotarod, and open-field tests, as described previously [16–18].
Estimation of hydrogen sulphide, dopamine, DOPAC, and marker enzymes
The estimation of hydrogen sulphide concentration in brain tissue and plasma samples was carried out as per the previous publications [19, 20] with few modifications per our tissue samples. This spectrophotometric method involves the reaction of sulphide with N,N-Dimethyl-p-phenylenediamine sulphate in the oxidizing agent Fe3+ in hydrochloric acid forming methylene blue that is read at 670 nm. Furthermore, brain tissue extract sample was analysed for dopamine (DA) and DOPAC (3,4-dihydroxyphenylacetic acid) by high-performance liquid chromatography (HPLC) fitted with a Shimadzu L-ECD-6A electrochemical detector coupled with a Shim-Pak CLC- ODS column (25 cm length), with flow volume of 0.5 ml/min and mobile phase comprised of 150 mM monohydrate citric acid, 67 mM sodium octyl sulphate, 2% tetrahydrofuran, and 4% acetonitrile in deionized water (pH 3.2). The analysts’ retention time was determined using the standard monoamines, test samples were quantified by comparison with standards, and the results are expressed as ng/g wet tissue. Further monoamine oxidase (MAO) was assayed using commercial kits as per the manufacturer’s instructions. Also, ELISA kits for α-synuclein and tyrosine-hydroxylase were obtained from Fine Biotech, China, and commercial assay kits to analyse HDAC and HAT activity were obtained from Epigentek, USA.
Reverse transcription-PCR
In order to evaluate the expression of microRNA in the present study, quantitative RT-PCR was performed with miR-specific primers. RNA was isolated from the brain tissues using Trizol reagent. For quantitative detection of miR-34b, miR-205, miR-433, miR-19b, and miR-124, TaqMan MicroRNA reverse transcription kit, TaqMan miR specific assays, and snoRNA assays were used according to the manufacturer’s instructions (Thermo Fisher Scientific, USA). SnoRNA was used as a control for normalization. To further elucidate the mRNA expression of PD markers, total RNA was extracted from the brain tissues using TRIzol® reagent. Briefly, brain tissues were homogenized in TRIzol and an equal amount of chloroform was added and centrifuged at 10,000 g for 15 min at 4°C. The top aqueous phase was collected in a separate tube and mixed with isopropanol and incubated for 10 min at 25°C and later centrifuged at 12,000 g for 10 min at 4°C. The obtained RNA pellet was washed with 70% ethanol, and the total RNA was quantified using a spectrophotometer. A known amount of RNA sample was transcribed to cDNA, and real-time PCR was done for specific genes using the SYBR® Green/ROX master mix (Qiagen). The gene-specific primers used in the study are listed in the Table I. The obtained Ct values were used to calculate gene expressions by the comparative Ct method (ΔΔCT) using GAPDH as the housekeeping gene.
Table I
List of primer details
Statistical analysis
Statistical significance was evaluated with a one-way analysis of variance (ANOVA) and Student’s t-test methods. Data were expressed as the mean ± SEM. Graph Pad Prism version 5.0 was used for the analyses. Differences with a p-value of less than 0.05 were considered statistically significant.
Results
To evaluate the protective role of HDAC inhibition in alleviating Parkinson’s disease (PD), the present study was conducted using the rat model of PD and administered with the HDAC inhibitor entinostat. Initially, the onset of PD was assessed using the neurological assessment, and the results demonstrated that significant reductions (p < 0.05) in the latency seen on the rotarod test apparatus were found in PD rats compared to the control group. In addition, the observed exploratory activity in the open field with anxious-like behaviour and depressive-like behaviour in PD-induced rats compared to controls. However, entinostat-administered rats restored the neurological functions in the PD rats (Figure 1).
Figure 1
A–D – Represents the neurological function test such as elevated plus maze test, forced swim test, Morris water maze, and motor performance in the control and experimental groups of rats, respectively
Values are expressed as mean ± S.E (n = 12). Statistical significance expressed as *p < 0.05, **p < 0.01 compared to vehicletreated controls, $p < 0.05 entinostat compared to PD rats.
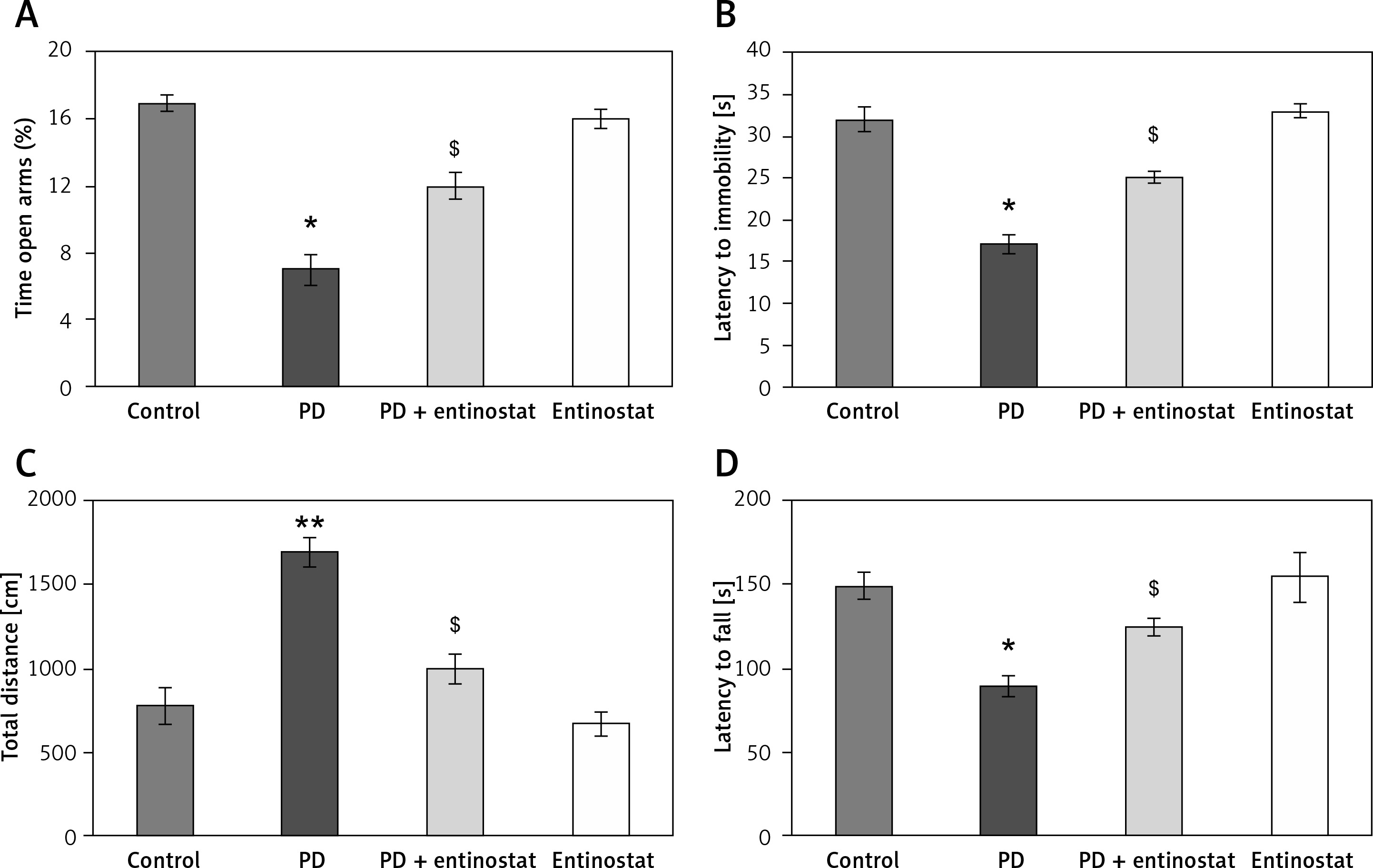
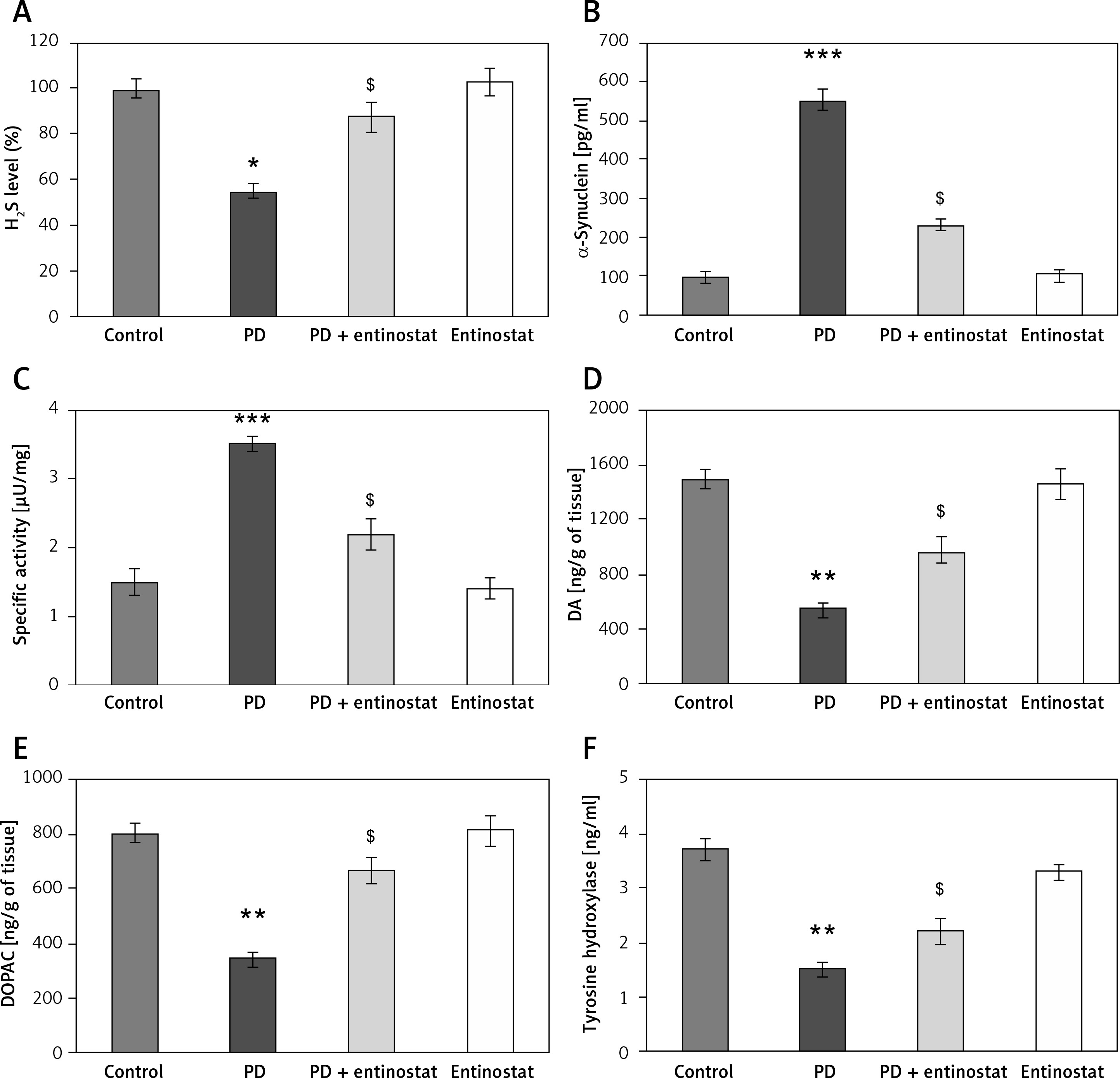
Figure 2 shows the effect of entinostat on the levels of H2S, and other metabolites of PD importance have been evaluated in control and PD-induced rats. The levels of H2S were found to be decreased in both the plasma and brain tissues of PD-induced rats compared to control rats. Furthermore, the reduced levels of dopamine (p < 0.05), 3,4-dihydroxyphenylacetic acid (DOPAC) (p < 0.05), and tyrosine hydroxylase (p < 0.05) with increased levels of α-synuclein and MAO activity and were observed in PD rats compared to controls. The administration of entinostat elevated the levels of H2S (p < 0.05) and restored the neurochemical equivalent to controls, suggesting protective effects of HDAC inhibition (Figure 2).
Figure 2
A–F – Represents the levels of H2S, DA, DOPAC, α-synuclein, and marker enzymes of control and experimental groups of rats, respectively. The amount of DA and DOPAC is expressed as ng/g tissue. The amount of H2S release was compared between groups and expressed as percentages
Values are expressed as mean ± SE (n = 12). Statistical significance expressed as *p < 0.05, **p < 0.01 compared to vehicletreated controls, ?p < 0.05 entinostat compared to PD rats.
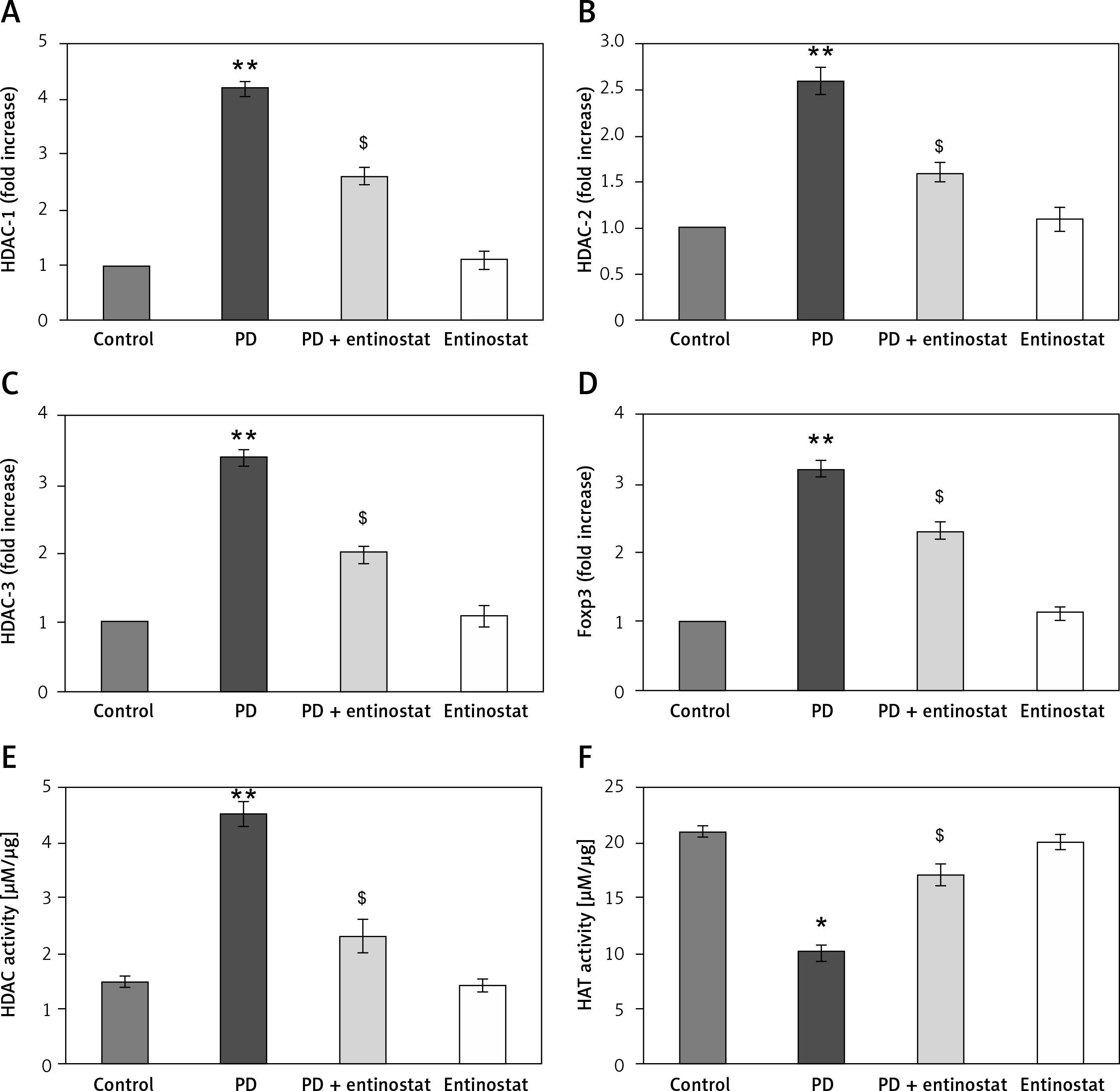
Moreover, to substantiate the role of HDAC inhibition on various HDAC genes classes, the mRNA levels of HDACs were identified by real-time quantitative PCR in relation to the control gene, and the results are shown in Figure 3. The results show a profound upsurge (p < 0.01) in the mRNA expression of Foxp3 (3.2-fold), HDAC-1 (4.2-fold), HDAC-2 (2.6-fold), and HDAC-3 (3.4-fold) with increased HDAC activity and reduced HAT activity in PD rats compared to controls. However, the increased levels of these HDAC genes were found to be reduced in entinostat treatment, indicating that the HDAC inhibition must be a major player in initiating the restorative mechanism in PD treatment (Figure 3).
Figure 3
A–F –Represents qRT-PCR mRNA expression analysis of HDAC genes in the control and experimental groups of rats. The fold increase of gene expression is compared with the housekeeping gene GAPDH. E, F – Total HDAC and HAT activity. The detail of the experiment is given in the methodology section
Values are expressed as mean ± SE (n = 12). Statistical significance expressed as *p < 0.05, **p < 0.01 compared to vehicletreated controls, $p < 0.05 entinostat compared to PD rats.
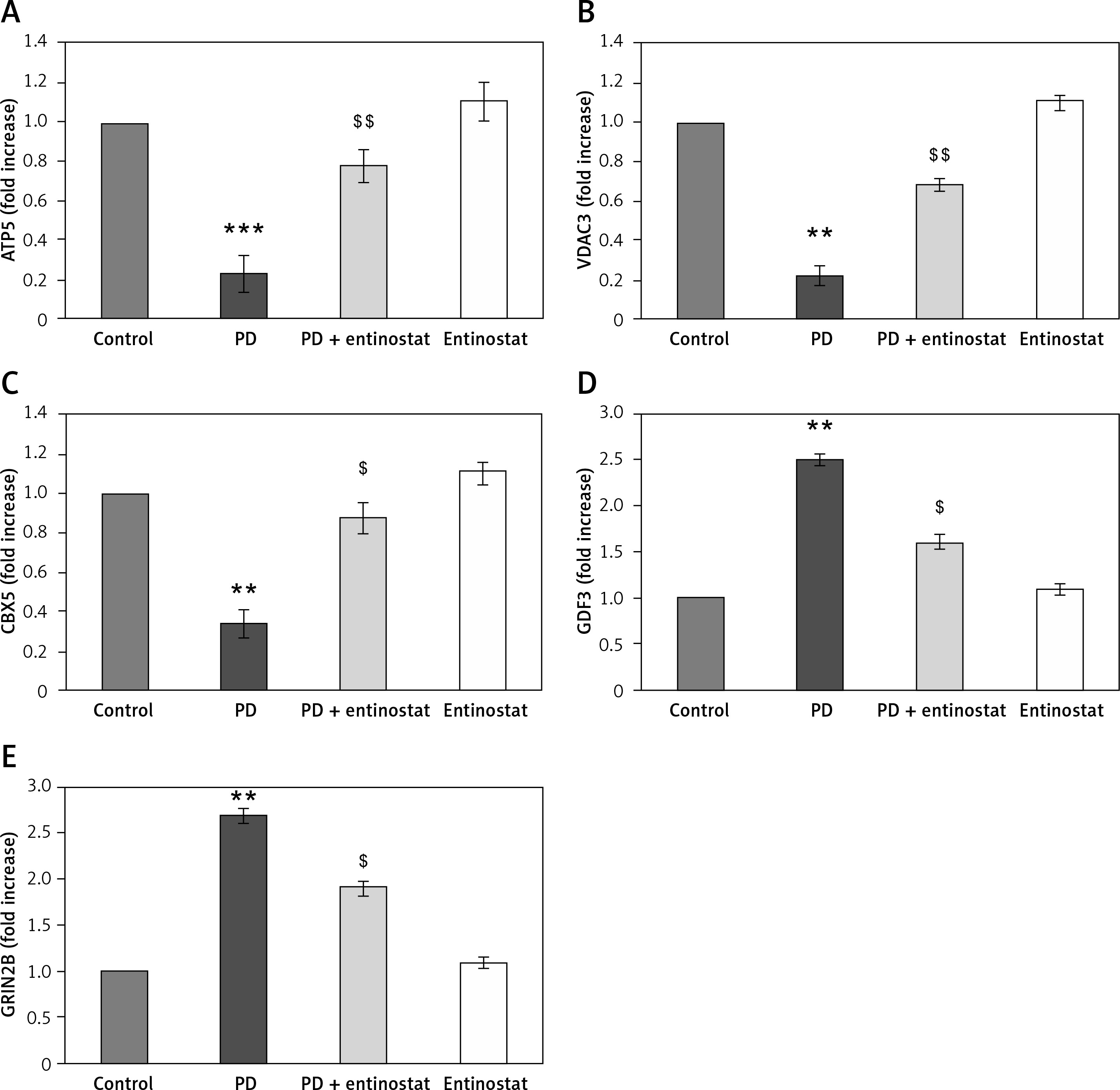
Conversely, an additional experiment illuminating the PD marker gene levels was carried out, and the results are presented in Figure 4. In the present study, rats induced with PD using rotenone demonstrated a significant decrease (p < 0.05) in the levels of mRNA such as ATP5A1 (-2.3-fold), VDAC3 (-2.2-fold), and CBX5 (-3.3-fold) and increased levels of GDF3 (2.5-fold) and GRIN2B (2.7-fold) compared to controls. However, the levels of the genes that were restored in the group of PD rats receiving the HDAC inhibitor entinostat suggest its direct effect in cellular mechanisms involved in PD (Figure 4).
Figure 4
A–E – Represents qRT-PCR mRNA expression analysis of marker genes in the control and experimental groups of rats. The fold increase of gene expression is compared with the housekeeping gene GAPDH. The detail of the experiment is given in the methodology section
Values are expressed as mean ± SE (n = 12). Statistical significance expressed as **p < 0.01, ***p < 0.001 compared to vehicle-treated controls, $p < 0.05, $$p < 0.01 entinostat compared to PD rats.
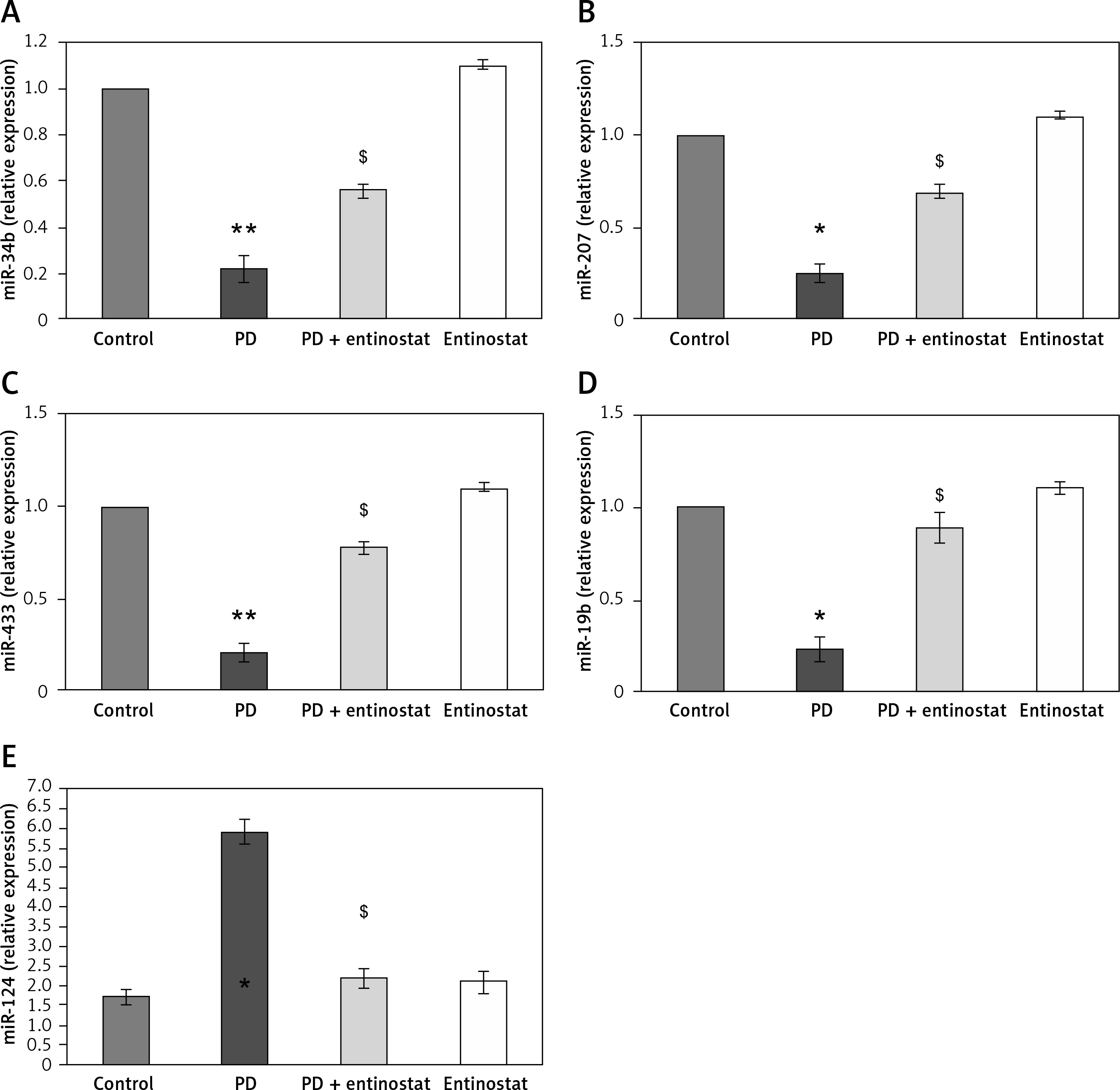
The role of HDAC signalling in PD was evaluated in terms of microRNA using qPCR. Figure 5 shows the expression level of miR-34b, miR-205, miR-433, miR-19b, and miR-124 in the control and experimental groups of rats. Rats induced with PD elicited a significant reduction (p < 0.05) in the expression of miR-34b (2.2-fold), miR-205 (1.7-fold), miR-433 (2.1-fold), miR-19b (2.3-fold), and miR-124 (2.6-fold). On the other hand, the levels of these miRs were restored in entinostat pre-treatment, suggesting that the interplay of microRNAs is one of the clues in the aggravation of PD (Figure 5).
Figure 5
A–E – Represents expression analysis of microRNAs such as miR-34b, miR-205, miR-433, miR-19b, and miR-124 in the control and experimental groups of rats, respectively. The detail of the experiment is given in the methodology section
Values are expressed as mean ± SE (n = 12). Statistical significance expressed as *p < 0.05, **p < 0.01 compared to vehicle-treated controls, $p < 0.05 entinostat compared to PD rats.
Discussion
In this study, we have discussed the putative role of HDAC in PD through the modulation of expression of various markers that are known to be associated with PD, and the potential role of entinostat as a new pharmacological molecule in the treatment of PD (Figure 6). H2S is produced endogenously under physiological conditions for its role in the brain physiology. Patients suffering from neurodegenerative diseases like Alzheimer’s [21] and PD [22] show decreased production of H2S in their brains. Our animal models showed that H2S is decreased in the PD-induced group, and it correlates highly with the neurodegeneration [23]. Treatment with entinostat not only improved the H2S production levels to near normalcy but also improved neuroprotection. With hydrogen sulphide providing neuroprotection to patients with Parkinson’s disease [23], treatment with entinostat would have enhanced the hydrogen sulphide production and hence exerted its neuroprotective effects [24].
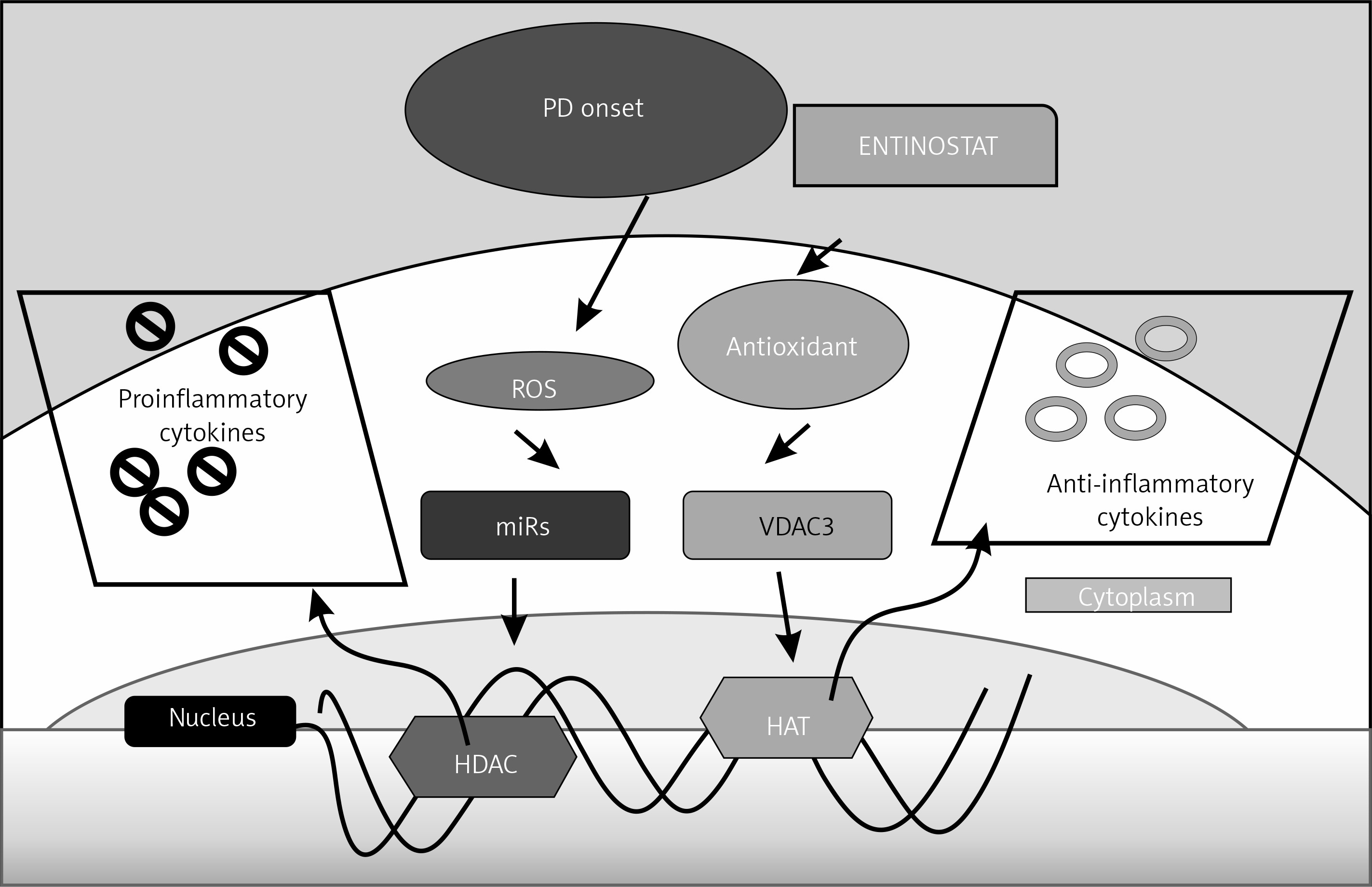
Figure 6
Schematic representation of the probable mechanisms by which entinostat exerts inhibition on cell dysfunction in PD. PD onset enhances ROS release activation with depletion of an antioxidant defence mechanism, which further activates the unregulated shuttling of proinflammatory cytokines with enhanced activation of HDAC with suppressed HAT activity. However, treatment with entinostat enhanced the antioxidant enzymes with regulated levels of deranged cytokines, and HDAC genes suggest that HDAC inhibition can ameliorate PD-mediated harmful cytokines
The level of expression of α-synuclein is directly correlated with the PD phenotype, and the accumulation of α-synuclein levels in the substantia nigra contributes to the PD pathogenesis [25]. In our models, the animals with the highest expression levels of α-synuclein were induced with PD. The action of entinostat decreased the expression of α-synuclein in the other successive groups. Similarly, the effect of PD is enhanced with increased activity of monoamine oxidase (MAO), which deaminates DA to generate a hydroxyl radical that contributes to the oxidative stress in PD [26]. MAO activity is increased in concordance with the increase in α-synuclein to increase the oxidative stress in PD-induced rats, which is reduced in entinostat-treated groups. The action of entinostat follows its counterpart vorinostat in being a HDAC class I inhibitor [9, 27] in counteracting the effect of an increase of α-synuclein by preventing its translocation to the nucleus for pathogenicity.
We assessed the ratios of levels of dopamine (DA): 3, 4-dihydroxyphenylacetic acid (DOPAC), which provides a biomarker of Parkinson’s disease in patients with established synucleinopathies. The plasma and brain tissues of our PD-induced animals showed a decrease in the ratio of both DA and DOPAC, which is a typical early diagnosis marker of Parkinson’s. The depletion of DA from the nigrostriatum indicates PD, and the DOPAC levels in CSF are also decreased in patients with movement disorders [28]. These decreases in the PD animals were reversed in animals that were treated with entinostat, which increased the levels of DA and DOPAC.
A similar increase in tyrosine hydroxylase levels, which is a rate-limiting enzyme in the biosynthesis of DA [29], has been observed. Parkinson’s disease is characterized by the loss of dopaminergic neurons and the loss of DA in the substantia nigra. The reduction in the levels of TH causes loss in DA synthesis, and this leads to PD. Thus, TH levels play a major role in the pathogenicity of PD. In cases where the synuclein increases, TH activity is compromised and decreases the DA synthesis, leading to PD [30]. The use of entinostat has effectively decreased the synuclein and increased the activity of TH to increase DA, and thereafter DOPAC synthesis, in the treated animals.
Histone deacetylases (HDACs) are enzymes that catalyse the removal of acetyl groups from lysine and arginine residues of amino-terminal tails of histones [31]. The removal of acetyl groups returned the chromatin to a condensed form that prevents the transcription of various genes from expression. The complete pattern of expression of HDACs would be ideal to find the targets for inhibiting them, and thus it could be used as a therapeutic strategy [12]. In order to find which of the HDAC isoforms are involved in the process of the pathophysiology of PD, we tested the mRNA levels of HDAC isoforms 1, 2, and 3 and found them to be increased in all of the PD-induced animal groups, as already proven here [32], and hence a narrow-spectrum HDAC inhibitor is needed. When tested our molecular candidate for HDAC inhibition, entinostat significantly reduced the mRNA expression levels of HDACs 1, 2, and 3 in all the treated groups [33].
The HDAC inhibiting activity of entinostat was substantiated by a significant decrease in the HDAC activity, as researched here [34], and a simultaneous increase in the HAT activity was observed in the animals of the entinostat-treated group. We, for the first time, used entinostat in the HDAC inhibition in PD. Furthermore, entinostat suppresses Foxp3 expression mRNA, leading to decreased Foxp3 protein, causing a defective impaired response on the Treg population to cause immunomodulation in PD [35]. These observations exhibited in our entinostat-treated group of animals are in line with established results, in which a similar decrease in the expression of Foxp3 at the transcription level was observed [35, 36].
Disruption of mitochondria and its metabolic activity are primarily focused on the pathogenesis of PD [37]. There is a dysfunction of mitochondria in neurodegenerative diseases utilizing very little glucose in the decreased ATP synthase activity with reduced energy production [38, 39]. These are known to be the hallmarks of neurodegenerative diseases. Accompanying this would be the decrease in ATP5A1 subunits that oxidative impairment has destabilized the protein as well as its enzymatic activity [40]. The ATP5A1 expression is severely compromised in the PD-animal group because the data fits well with the established results of neurodegeneration and subsequent impairment of brain metabolism [39]. The expression and activity are restored with the action of entinostat in the treated group due to its mitochondria preservative activity and by maintaining ATP [41, 42]. The pathologies of VDAC isoform 3 can be attributed to its altered levels of gene expression [43, 44]. Conversely, the present study results also reflect decreased isoform 3 expression in the PD-animal group and returned to near normalcy in the entinostat-treated group.
Much of the gene expression profiling already performed to differentiate between the PD patients and healthy controls identifies ATP synthase subunit α (ATP5A1), voltage-dependent anion-selective channel protein 3 (VDAC-3) [45], growth differentiation factor 3 (GDF3) [46], glutamate receptors, glutamate ionotropic receptor NMDA type subunit 2B (GRIN2B) [47], chromobox homologue 5, and CBX5, confirmed by qRT-PCR to be found in the substantia nigra dopaminergic neuron degeneration [48] or acting upon mitochondria in the pathogenesis of PD. More studies are needed to elucidate the exact roles that they play in PD animals beyond the scope of PD marker identification.
Small non-coding RNAs, miRs, bind to the 3’- UTR of the mRNA and regulate the gene expression, and dysregulation of miR expression is associated with PD and other neurodegenerative diseases [49, 50]. Hence, we explored the miRs that are involved in PD pathogenesis. The importance of miR-34b in PD pathogenesis is known due to the fact that it has low expression in parts of the brain in PD patients [51]. When the expression of miR-34b is reduced, oxidative stress increases with decreased mitochondrial metabolism and decreased viability of dopaminergic neuronal cells. Also, its inhibition can increase the α-synuclein levels towards PD pathogenesis [52]. Treatment with entinostat restored the cell viability, mitochondrial function, and downregulation of ROS in the animals. The expression of miR-433 was found to be decreased in PD patients’ serum and CSF and is in line [34] with our results in which a decrease in the expression of miR-433 is observed in PD-induced animals. The increase in alpha-synuclein or under conditions of synucleinopathies, miR-19b, was known to be downregulated and also serves as a biomarker for PD. These increases in pathogenic miR have been countered by the corresponding increase in miR-124, which is highly expressed in the brain and provides a neuroprotective role in PD, which is highly expressed in entinostat-treated animals (Figure 6). More studies are required to decode the mechanism by which entinostat reduces Parkinson’s miR markers from their effect on the physiology of the disease. The results obtained are not specific because these markers are well known as biomarkers of PD.
In conclusion, our study results elucidate partial molecular mechanisms by which the PD animals relieve their neurodegenerative symptoms and restore their coordinated movements with the rehabilitation of mitochondria that are the primary site of pathogenesis in PD, as well as by alleviating the biomarkers of PD by its primary action on HDAC. Entinostat would thus serve as a general and an epigenetic regulator against HDACs. Further mechanisms by which this molecule would act require a more systematic approach involving recent genetic findings and would be a better neuroprotective mediator of animal models with PD.