Haemoglobinopathies constitute the most common group of hereditary disorders in humans; its inheritance pattern is autosomal recessive, and the pathophysiology is related to the abnormal structure or synthesis of haemoglobin [1]. Sickle cell disease is caused by a point mutation in the first exon of the β-globin gene (HBB) with the substitution of adenine for thymine and the consequent exchange of glutamic acid by valine at the 6-position (β6Glu→Val) of the globin chain, which changes the molecular structure of haemoglobin and produces haemoglobin S (Hb S) [2]. At low oxygen concentrations Hb S polymerises, resulting in the deformation of erythrocytes, which adopt an inflexible sickle shape and alter the viscosity of the blood, the immediate cause of vascular occlusion and anaemia [2, 3]. In homozygosis, the Hb S mutation characterises sickle-cell anaemia (Hb SS), the most common and severe condition of the disease, whereas in heterozygosis it characterises asymptomatic sickle cell trait (Hb AS) [4]. Other genotypes can occur when there is an interaction of the Hb S mutation with other variants related to the β-globin gene, such as haemoglobin (Hb) C, Hb E, and Hb D, or with β-thalassemia genotypes (HbS-β) [5].
Beta-S globin haplotypes (ßS haplotypes) are a set of polymorphisms in the HBB gene family, identified with specific enzyme restriction sites and named according to their region of origin and prevalence: Bantu (CAR, South Africa, Central Africa), Benin (BEN, Central West Africa), Senegal (SEN, West Atlantic), Cameroon (CAM, West Coast Africa), and Arabic-Indian (SAUDI, Indian subcontinent, East Arabian Peninsula) [6]. These are important markers for understanding the origin and evolution of the mutations for Hb S and the phenotypic expression of sickle cell disease (SCD) [7, 8].
In turn, glucose-6-phosphate dehydrogenase (G6PD) is a cytoplasm metabolic enzyme that catalyses the first step in the phosphate pentose pathway and leads to the reduction of NADP+ to NADPH, which continuously participates in the glutathione cycle (GSH), which protects red cells against oxidative damage and prevents acute haemolytic anaemia [9, 10]. Deficiency of G6PD (G6PDd) is a genetic disease, linked to the X chromosome, which has more than 400 different biochemical variants described due to the genetic heterogeneity of the gene locus, and caused by mutations that result in substitutions or single deletions of amino acids [11]. The variant G6PD A- (G202A/A376G) occurs with the change of guanine by an adenine at the nucleotide position 202, which causes a chain substitution of valine by methionine at the amino acid position 68, in combination with an adenine substitution by guanine at nucleotide 376, which alternates asparagine with aspartate at amino acid position 126. This is an African originated mutation more commonly reported in the American continent, which represents only 8–20% of activity considered normal for the enzyme [10, 12].
Thus, due to the composition of the Brazilian population, the frequency of haemolytic anaemia and the importance of the screening and characterisation of this anaemia for public health, a case with the mutations for Hb S and G6PDd concomitantly is described, which enables the understanding of the possible origin of the mutations and explanation for the clinical aspects.
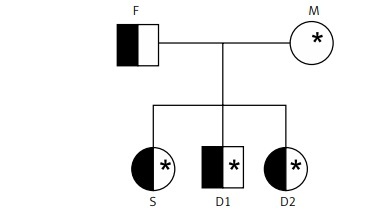
A 25-year-old male individual reported anaemic crises during childhood and volunteered to participate in a study on the frequency of G6PD deficiency and haemoglobinopathies performed by the Laboratory of Haemoglobin and Genetics of Haematological Diseases from UNESP – São José do Rio Preto, São Paulo, Brazil. After screening tests for haemoglobinopathies and G6PD deficiency (G6PDd), he was diagnosed with sickle cell trait (Hb AS) and G6PDd caused by G6PD A- variant. Then, his family volunteered to perform the same tests to understand the family history and the origin of the mutations. The family is formed by the mother (M) – 47 years old, father (F) – 50 years old, 2 daughters – (D1) 27 years old and (D2) 20 years old, and a son (S) – 25 years old.
Peripheral blood samples of all individuals of the family were collected after informed consent, following the Brazilian ethics legislation (Ethics Committee approval number 20123.413.0.0000.0019), and stored in tubes with ethylenediaminetetraacetic acid (EDTA) as an anticoagulant. The samples were submitted to classical tests for diagnosis of haemoglobinopathies, and the qualitative method of Brewer [13] was used for G6PD screening, all within 24 h of sample collection, to avoid interference of sample degradation in the enzymatic activity. The classical tests for the screening of haemoglobinopathies included analysis of the erythrocyte morphology [14], osmotic globular resistance in 0.36% sodium chloride solution [15], haemoglobin electrophoresis at alkaline pH (8.6) [16], haemoglobin electrophoresis at acid pH (6.8) [17], and haemoglobin profile and quantitative fractions by high-performance liquid chromatography (HPLC – Ultra2 – Resolution, TRINITY Biotech).
The DNA of the samples was extracted from the leukocytes by the salting-out method [18]. Molecular analysis of the haemoglobin mutations (Hb S, Hb C, Hb D, and β-thalassaemia), mutation investigation for G6PD, G202A (Hin1II) [19], and A376G (BseGI) [20], and determination of the Beta-S gene haplotypes (XmnI, HindIII, HincII, HinfI) [21] were performed by the PCR-RFLP method, as previously described.
After the classical haemoglobinopathy tests and the qualitative method of Brewer [13], we confirmed, by chromatography and molecular analysis, the presence of sickle trait (Hb AS) in the father and all the children. Also, the G6PDd (variant G6PD A-G202A/A376G) was found in the mother (heterozygous GA/AG) and one of the daughters (heterozygous GA/AG). The G6PDd was also found in the son (hemizygous A/G). The family descent history can be visualised in the heredogram in Figures 1 and 2.
Figure 1
Heredogram illustrating family descent history (the abbreviations do not correspond to the original name of the patients)
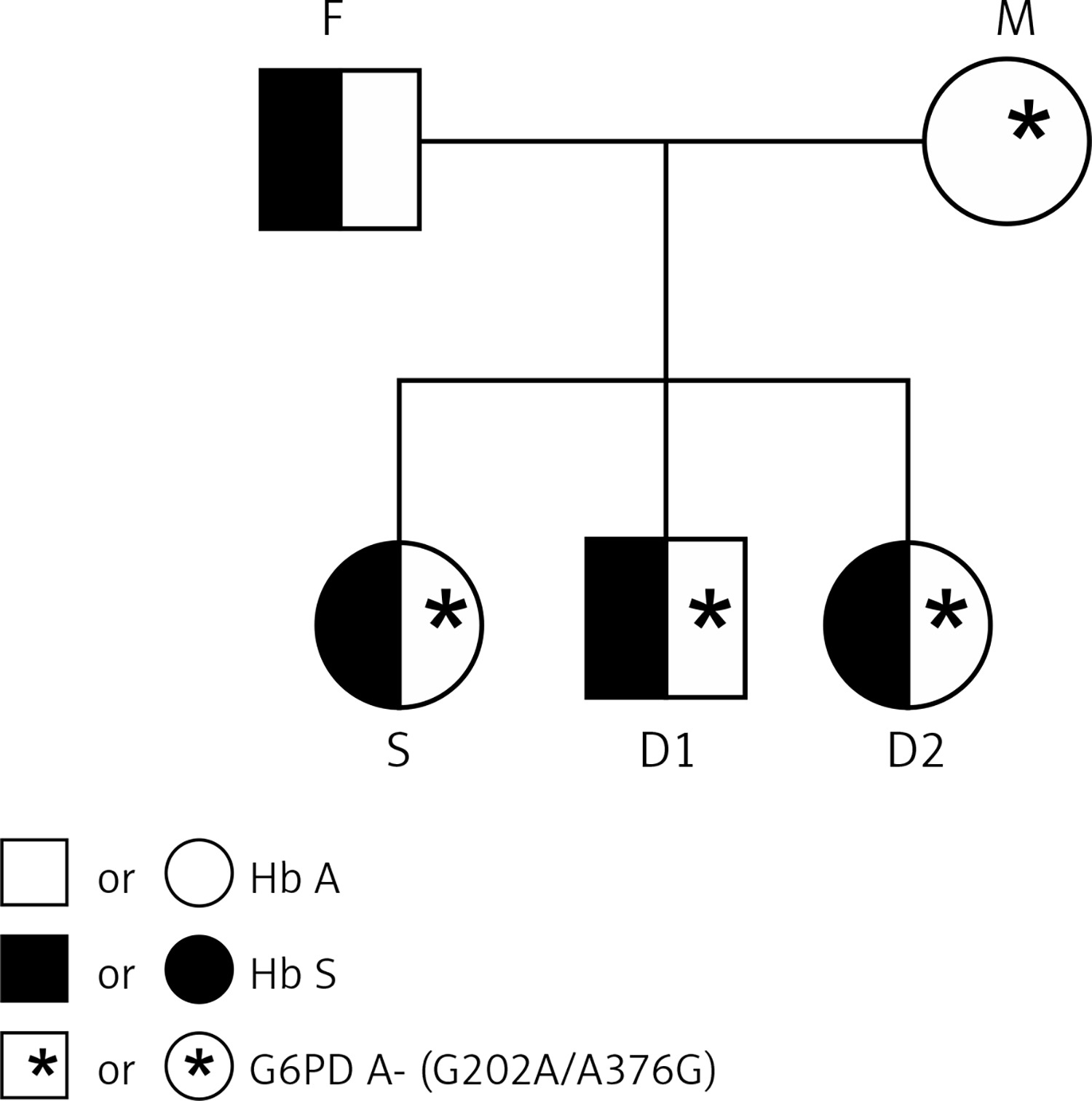
Figure 2
Agarose gel 3% showing: 1. Deficient homozygous sample for the SNP G202A (AA/A) – 63 and 46 bp fragments; 2 and 3 – Heterozygous sample for the SNP G202A (GA) – 109, 63 and 46 bp fragments – normal homozygous sample for the SNP G202A (GG/G) – non-digested 109 bp fragment
pb: base pairs; P.M.: ladder
In beta-S haplotypes analysis, the following haplotype patterns were found: Mother – Senegal/Atypical; Father – Bantu/Cameroon; and all children: Bantu/Senegal. They are summarised in Table I.
Table I
Summary of mutations and haplotype patterns found in the family’s genome
| Volunteers* | Hb | G6PD A- (G202A /A376G) | ßS Haplotype |
|---|---|---|---|
| F | AS | G/A | Bantu/Cameroon |
| M | AA | GA/AG (G6PD A-) | Senegal/Atypical |
| D1 | AS | GG/AA | Bantu/Senegal |
| S | AS | A/G (G6PD A-) | Bantu/Senegal |
| D2 | AS | GA/AG (G6PD A-) | Bantu/Senegal |
The results of screening tests for mutations in the family showed that the offspring inherited the father’s sickle cell trait while the mother, with a normal haemoglobin profile (Hb AA), transmitted the G6PDd A- mutations for 2 of the children. First, the fact that patients suffer from anaemic crises during childhood could be linked to environmental factors such as lack of basic sanitation, poor diet, and worms. The haplotype patterns found agree with what is expected in the Brazilian population, because the Bantu and Senegal haplotypes are associated with the Eastern and Western Atlantic regions of Africa, respectively, where there was intense forced migration of Africans to Brazil historically [6]. In contrast, the Cameroon haplotype is uncommon in Brazil because it is associated with a small region on the African West Coast. Furthermore, the presence of atypical haplotypes in the Brazilian population is explained due to the great admixture of the population and by several genetic events such as point mutations and recombination chromosomes that occurred more than 500 years ago in the country [8].
The Bantu haplotype is related to greater clinical severity of sickle cell disease, whereas the Senegal haplotype is related to low clinical complications [22]. These haplotypes were observed in the couple’s son, M.R.S., in whom severe anaemia had been reported throughout childhood, requiring blood transfusions, and who was only diagnosed with Hb S and G6PD deficiency co-inheritance.
In addition, although patients with G6PDd are asymptomatic, when in contact with oxidizing agents, they have risks of damage and intense anaemia; thus, access to this information is extremely relevant for patients to avoid clinical complications. According to the patient’s manual on Hereditary Hemolytic Anemia of HEMORIO (Institute of Hematology Arthur de Siqueira Cavalcanti, Rio de Janeiro, Brazil), oxidative damage to erythrocytes is caused by the use of high doses of certain drugs, like analgesics, antibacterials, arrhythmics, antihypertensives, and even ingestion of vitamins such as ascorbic acid and vitamin K. Also, contact with oxidising substances such as naphthalene, dyes used in some processed foods, ingestion of fava beans, and bacterial and viral infections [23] pose a risk of oxidative damage to erythrocytes.
In conclusion, although the individuals analysed have sickle cell traits or have normal haemoglobins, the study significantly contributed to the accurate diagnosis, prognosis, and genetic guidance regarding G6PD deficiency and coherence with haemoglobinopathies. Studies with the Bantu haplotype are related to the greater clinical severity of sickle cell anaemia. In this case, even though HbAS is a promoter, the association with hemizygosity of G6PDd and inherited haplotypes may contribute to a greater susceptibility to haemolytic events. Proper guidance can guarantee a better quality of life and access to preventative measures, as well as information for future reproductive decisions.