Introduction
Endometrial carcinoma is the most common gynaecological malignancy, currently representing 13% of all female cancers [1, 2]. The American Cancer Society estimates that there were 43,470 new cases and 7950 deaths from endometrial cancer in 2010 [3]. Current therapies for advanced and recurrent endometrial cancer remain ineffective.
Dietary n-3 polyunsaturated fatty acids (PUFAs) inhibit the growth of endometrial cancers, which have been identified as the most common hormone-responsive tumours both in vitro and in vivo [4, 5]. Epidemiological data suggest an inverse relationship between endometrial cancer risk and consumption of a diet rich in PUFAs, including docosahexaenoic acid (DHA, 22:6n-3) and eicosapentaenoic acid (EPA, 20:5n-3) [6, 7]. DHA supplementation is negatively correlated with cancer cell proliferation and tumour development in many cancer types. Importantly, while DHA selectively inhibits such proliferation, it is significantly less toxic towards normal cells [8–10]. It has been suggested that DHA induces apoptosis by regulating mitochondrial-related mechanisms at the membrane [11]. Studies have demonstrated that the incorporation of DHA into mitochondrial membranes increases susceptibility to reactive oxygen species (ROS) damage generated in colonocytes [12] and the HT-29 human colon tumour cell line [13]. Specifically, the association between DHA and cardiolipin, a mitochondrial inner membrane phospholipid, contributes to a mitochondrial-dependent apoptotic pathway that initiates the release of cytochrome c from the mitochondrial intermembrane space, which is associated with the induction of oxidative stress, loss of mitochondrial membrane potential, and apoptotic signalling [14–16]. Two major apoptotic pathways have been proposed in normal human cells. The extrinsic apoptotic pathway involves the stimulation of death receptors including Fas and tumour necrosis factor receptor-1 and involves the adapter protein Fas-associated death domain, as well as the initiator caspase-8. In parallel, the intrinsic pathway of apoptosis results from changes in mitochondrial status, activation of the apoptosome, and release of mitochondrial cytochrome c into the cytosol, which initiates a caspase activation cascade [17, 18]. Activated caspase-8 in these systems directly activates the executioner caspase (caspase-3) or cleaves BID (a BH3-only member of the Bcl-2 family of proteins), which triggers its translocation to the mitochondria followed by the release of cytochrome c, Smac/DIABLO, and apoptosis inducing factor [19, 20]. Other studies have reported that pro-caspase-8 is translocated to the mitochondria, where it binds to cardiolipin, oligomerises, and is activated, leading to cleavage of truncated p15 BID (tBID) and the generation of proteolytically active p43 and p10 fragments [21].
An immunofluorescence analysis revealed that active caspase-8 and tBID are primarily located in the mitochondria, although a distinct portion of pro-caspase-8 also co-localised with tBID at the mitochondria following exposure to DHA and triacsin C. Therefore, it seems plausible that DHA and triacsin C induce translocation of caspase-8 to the mitochondria, where it binds BID, leading to activation of tBID in the mitochondria. In addition, a sub-G1 analysis revealed that DHA and triacsin C-induced apoptosis are completely suppressed by inhibition of caspase-8, indicating that apoptosis requires activation of caspase-8 and BID. Although these results show that apoptosis induced by DHA and triacsin C is involved in both the extrinsic and intrinsic apoptotic pathways, activation of caspase-8/Bid appears to play the most critical role in the induction of apoptosis in RL95-2 cells. Cumulative evidence has demonstrated the apoptotic effect and cytotoxicity of DHA against tumour development in many cell types [22, 23], but the precise cellular and biochemical mechanisms of DHA-induced apoptosis in human endometrial cancer cells have not been investigated.
Long-chain unsaturated fatty acids are normally processed by acyl-coA synthetases (ACSs). Acyl-coA synthetases catalyse the initial step of mammalian fatty acid metabolism by converting long-chain fatty acids to acyl-CoA, which function mainly as activated lipid intermediates for fatty acid β-oxidation, modulating cellular signal transduction [24, 25]. Studies with triacsin C have shown that inhibiting ACS leads to apoptosis in human T lymphoblastoma [26], glioma [27, 28], and hepatocellular carcinoma cells [29] through pathways mediated by mitochondria. Another study showed that treatment with simvastatin, a lipid-lowering drug, decreases proliferation of human acute monocytic leukaemia (THP-1) macrophage cells [30]. Based on these findings, it has been hypothesised that the inhibition of ACS by triacsin C would impair DHA metabolism, which contributes to cytotoxicity-induced apoptosis by DHA in RL95-2 endometrial carcinoma. Therefore, combining DHA with triacsin C might maximise the effect of apoptosis in endometrial cancer cell line.
In the present study, we evaluated whether triacsin C cooperatively induces apoptosis with DHA in RL95-2 cells. Then, we investigated whether the synergistic effects of triacsin C and DHA on apoptotic induction were mediated by changes in mitochondria accompanied by activation of caspase-8, -9, -7, and -3.
Material and methods
Cell culture
RL95-2 cells (human endometrial adenocarcinoma cell line; American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium nutrient mixture with HAMS F-12 (Sigma, St. Louis, MO, USA) containing 10% foetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 1.2 g/l sodium bicarbonate supplemented with 10 μg/ml penicillin-streptomycin (Invitrogen). The cells were incubated in a humidified incubator at 37°C with 5% CO2 and exposed to DHA or triacsin C when confluency reached 20%.
MTT cell viability assay
Cells were seeded in 12-well plates at a density of 5 × 105 cells per well. After an appropriate treatment time (24 or 48 h) with DHA and triacsin C, the culture medium was removed and replaced with a medium containing 0.5 mg 3-[4,5-dimethylthiazol-2-yl]-2.5-diphenyltetrazolium bromide; thiazolyl blue (MTT; Sigma) dissolved in phosphate-buffered saline (PBS, pH 7.2). After 4 h, the crystals were dissolved with 200 μl DMSO (Sigma). The intensity of the colour in each well was measured at a wavelength of 490 nm using a microplate reader (EL-312e; Biotek, Winooski, VT, USA).
Flowcytometric cell death assay
The cells were harvested, fixed in 95% ethanol for 24 h, incubated with 0.05 mg/ml propidium iodide (PI) and 1 μg/ml RNase A (Sigma) at 37°C for 30 min, and analysed by flow cytometry using Epics XL and analytic software (EXPO32 TM; Beckman Coulter, Brea, CA, USA). The cells belonging to the sub-G1 population were considered apoptotic cells; the percentage of each phase of the cell cycle was determined.
Hoechst 33258 staining
The cells were stained with Hoechst 33258 (4 μg/ml) for 30 min at 37°C, fixed for 10 min in 4% paraformaldehyde (PFA; Sigma), and observed under an Axiophot microscope (Carl Zeiss, Zena, Germany).
Annexin V cell death assay
The cells were stained using an Annexin V-FITC Apoptosis Detection kit (BD Biosciences, Parsippany, NJ, USA) according to the manufacturer’s instructions. The stained cells were analysed by flow cytometry.
Western blot analysis
Whole-cell lysates were prepared by incubating the cell pellets in lysis buffer (30 mM NaCl, 0.5% Triton X-100, 50 mM Tris-HCl (pH 7.4), 1 mM Na3VO4, 25 mM NaF, and 10 mM Na4P2O7) for 30 min on ice. After the insoluble fractions were removed by centrifugation at 14,000 rpm at 4°C for 30 min, the supernatants were collected, and protein concentration was determined with a BCA protein assay kit (Pierce Biotechnology, Woburn, MA, USA). The same amount of protein (~30 μg) was subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane. The membranes were incubated for 1 h at room temperature (RT) with a primary antibody in Tris-buffered saline containing 0.05% Tween-20 (TBS-T (pH 7.4)) in the presence of 5% nonfat dry milk. After the membranes were washed in TBS-T, secondary antibody reactions were performed with an appropriate antibody source conjugated with horseradish peroxidase. The signals were detected with an enhanced chemiluminescence detection kit (Amersham Pharmacia Biotech, Piscataway, NJ, USA) in an LAS-3000 detector (Fujifilm, Tokyo, Japan). Immunoblotting for β-actin was performed in every experiment as an internal control. All antibodies and reagents are summarised in Table I.
Table I
Reagents and antibodies
Immunocytochemistry
Harvested cells were attached on slide glasses by Cytospin centrifugation. The cells were fixed in 4% PFA, washed with PBS, and incubated with 0.2% Triton X-100. The cells were incubated with anti-cleaved caspase-3 and caspase-7 (Cell Signaling Technology, Beverly, MA, USA) primary antibodies in 1% bovine serum albumin at RT. The cells were incubated with FITC-conjugated anti-rabbit IgG and anti-mouse IgG, Texas Red-conjugated anti-rabbit IgG, and anti-mouse IgG (Vector Laboratories, Burlingame, CA, USA), and Alexa-conjugated anti-rabbit IgG (Invitrogen) secondary antibodies at RT. The cells were incubated with PI (50 μg/ml) at RT to counterstain the nucleus, if required. Finally, the cells were mounted and observed under a confocal microscope (LSM510; Carl Zeiss).
Results
DHA or triacsin C alone does not induce cell death in RL95-2 endometrial carcinoma cells
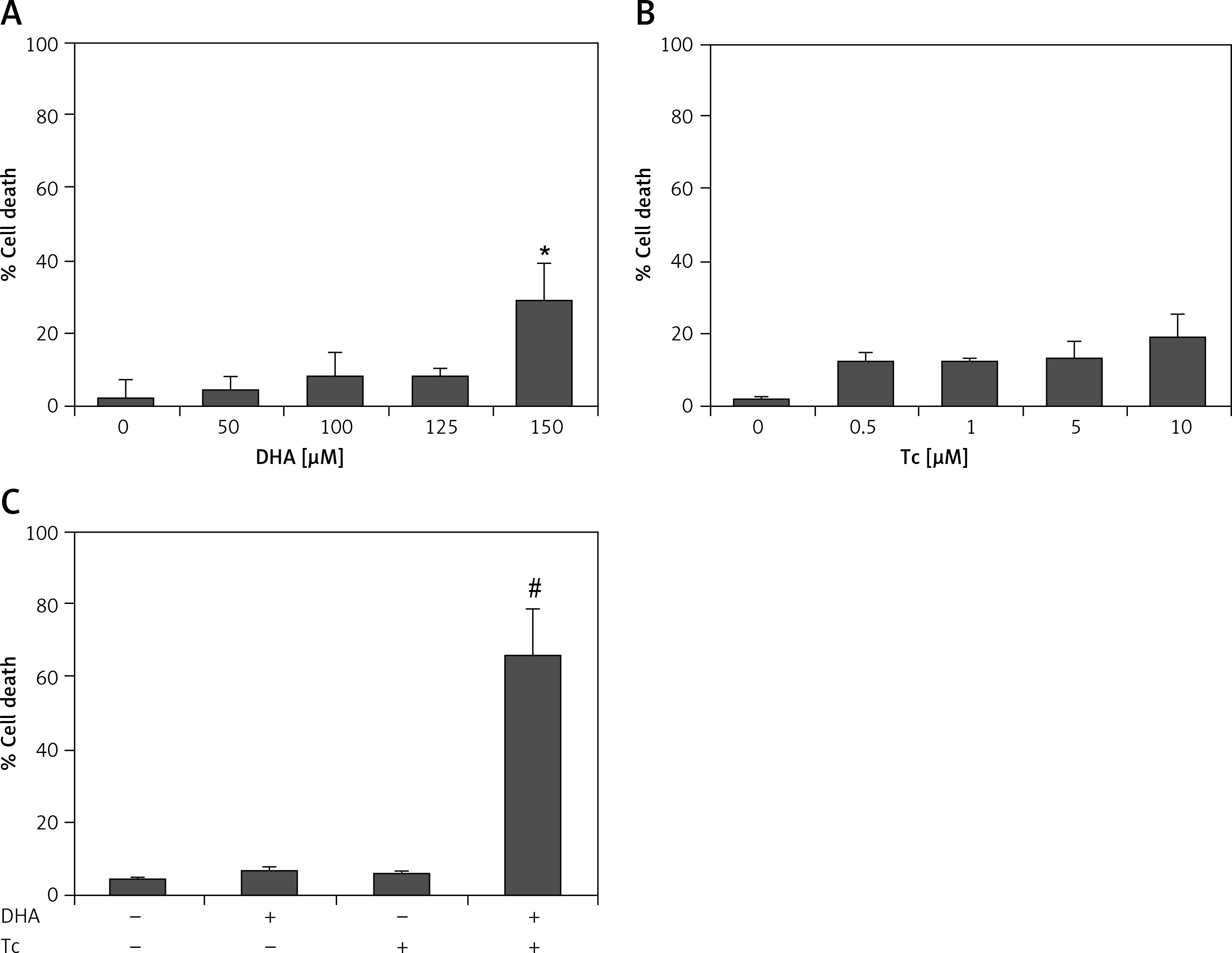
To evaluate the cytotoxic effects of DHA and triacsin C, RL95-2 cells were exposed to various concentrations of DHA (0–150 μM) or triacsin C (0–10 μM), respectively. Cell survival was estimated after 24 h of exposure to increasing drug concentrations using MTT and sub-G1 assays (PI staining). Cell viability was slightly reduced after increasing the concentrations of DHA or triacsin C (Figures 1 A, B). However, cell death as determined by sub-G1 analysis revealed that treatment with DHA or triacsin C alone did not induce significant cell death after 24 h at any of the concentrations tested (Figures 2 A, B).
Figure 1
Effect of docosahexaenoic acid (A) or triacsin c (B) on cell viability in RL95-2 endometrial cancer cells. Cell viability shown in cells treated with various concentrations of docosahexaenoic acid (0–150 μM) or triacsin c (0~10 μM) for 24 h and determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. DHA and Tc indicate docosahexaenoic acid and triacsin c, respectively
At least 3 independent experiments were performed, and data shown are the mean ± SD. *, p < 0.05 compared to 0 μM control.
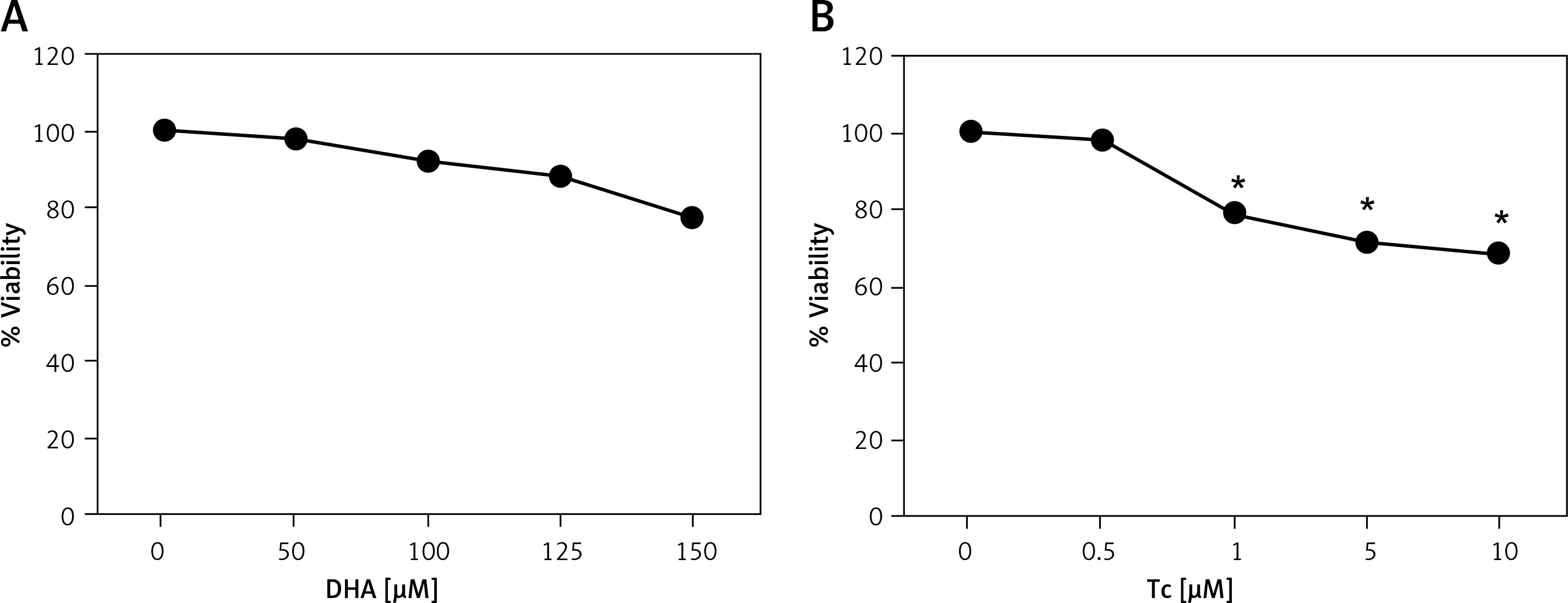
Figure 2
Effect of DHA or triacsin C on cell death in RL95-2 endometrial cancer cells. A – Cell death of cells treated with various concentrations of DHA (0–150 μM) for 24 h and assessed by flow cytometry is shown. Effect of triacsin C on cell death in RL95-2 endometrial cancer cells. B – Cell death of cells treated with various concentrations of triacsin C (0–10 μM) for 24 h and assessed by flow cytometry is shown. C – Effect of combination treatment with DHA and triacsin C on cell death in RL95-2 endometrial cancer cells. Cells were treated with 125 μM DHA, 5 μM triacsin C, or a combination of 125 μM DHA and 5 μM triacsin C for 24 h. DHA and Tc indicate docosahexaenoic acid and triacsin C, respectively. The percentage of cells with a sub-G1 DNA content was taken as a measure of cell death
At least 3 independent experiments were performed, and data shown are the mean ± standard deviation. *p < 0.05 compared to 0 μM control. #p < 0.001 compared with control, DHA-treated, or triacsin C-treated group.
Combination treatment with DHA and triacsin C significantly induces apoptosis in RL95-2 endometrial carcinoma cells
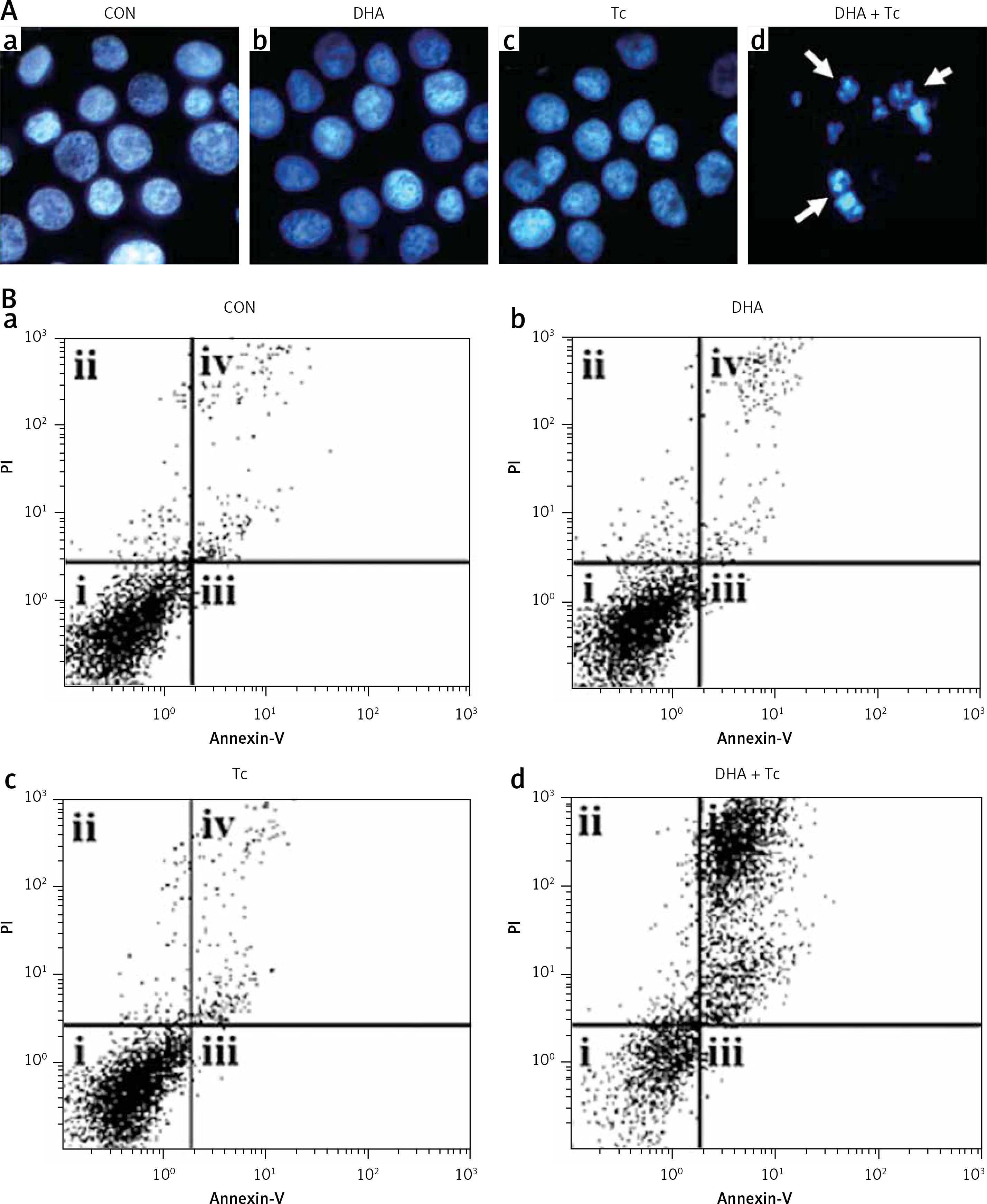
Figures 2 A, B shows that treatment with DHA or triacsin C alone was insufficient to induce cell death in RL95-2 cells. Based on these results, the optimal concentrations to induce cytotoxicity with combined treatment were determined in a series of combination experiments (data not shown): DHA concentration of 125 μM and triacsin C at 5 μM. Figure 2 C shows that after RL95-2 cells were exposed to combined treatment with 5 μM triacsin C and 125 μM DHA for 24 h, cell death increased significantly to 65.9 ±12.6% (p < 0.001). Hoechst staining of the cells exposed to combined treatment revealed nuclear fragmentation (Figures 3 A–D), while Annexin V staining revealed migration of the viable cell population (region i) toward late apoptosis (region iv) during treatment (Figures 3 B). These results suggest that the combined treatment of DHA and triacsin C synergised to induce cell death, which did not occur with treatment of DHA or triacsin C alone.
Figure 3
Nuclear fragmentation in RL95-2 endometrial cancer cells following exposure to combined treatment with DHA and triacsin C (A). Cells were treated with 125 μM DHA, 5 μM triacsin C, or a combined treatment of 125 μM DHA and 5 μM triacsin C for 24 h. Nuclear morphology was detected by Hoechst 33258 staining. Arrows point to fragmented nuclei. Original magnification: ×800. The proportion or type of cell death was identified by the Annexin V cell death assay. i, ii, iii, and iv denote viable (live), necrotic, early apoptotic, and late apoptotic regions, respectively (B)
DHA and triacsin C-induced apoptosis initiates through caspase-8/tBID and subsequently activates caspase-9, -3, and -7
To clarify whether DHA and triacsin C-induced apoptosis involves caspase activity, activation of the initiator (-8 and -9) and effector (-3 and -7) caspases was analysed by western blot and immunocytochemistry. Anti-procaspase-8, procaspase-9, proBID, and tBID were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-cleaved caspase-8, -9, -3, and -7 were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-poly ADP ribose polymerase (PARP) was purchased from Calbiochem (San Diego, CA, USA). Anti-XIAP was obtained from BD Pharmingen (San Diego, CA, USA). Anti-rabbit and mouse Ig-conjugated with horseradish peroxidase were purchased from Amersham Pharmacia Biotech. As shown in Figures 4 and 5, combined treatment with DHA and triacsin C invoked activation of caspases-3 and -7 as well as cleavage of PARP and XIAP (Figure 4). In addition, consistent with the western blot results, increased levels of caspase-3 and -7 were observed in DHA and triacsin C-treated RL95-2 cells (Figure 5).
Figure 4
Activation of caspase-3 and -7, PARP cleavage, and XIAP in RL95-2 endometrial cancer cells by combined treatment with DHA and triacsin C. Cells were treated with 125 μM DHA, 5 μM triacsin C, or a combination of 125 μM DHA and 5 μM triacsin C for 24 h. A – Activation of caspase-3 and -7 following exposure to DHA and triacsin C, as shown by western blot analyses for caspase-3 and -7, and PARP cleavage. Equal amounts of protein (30 μg) were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and immunoblotted using cleaved caspase-3, cleaved caspase-7, PARP, and XIAP antibodies, respectively. Actin was used as an internal (loading) control
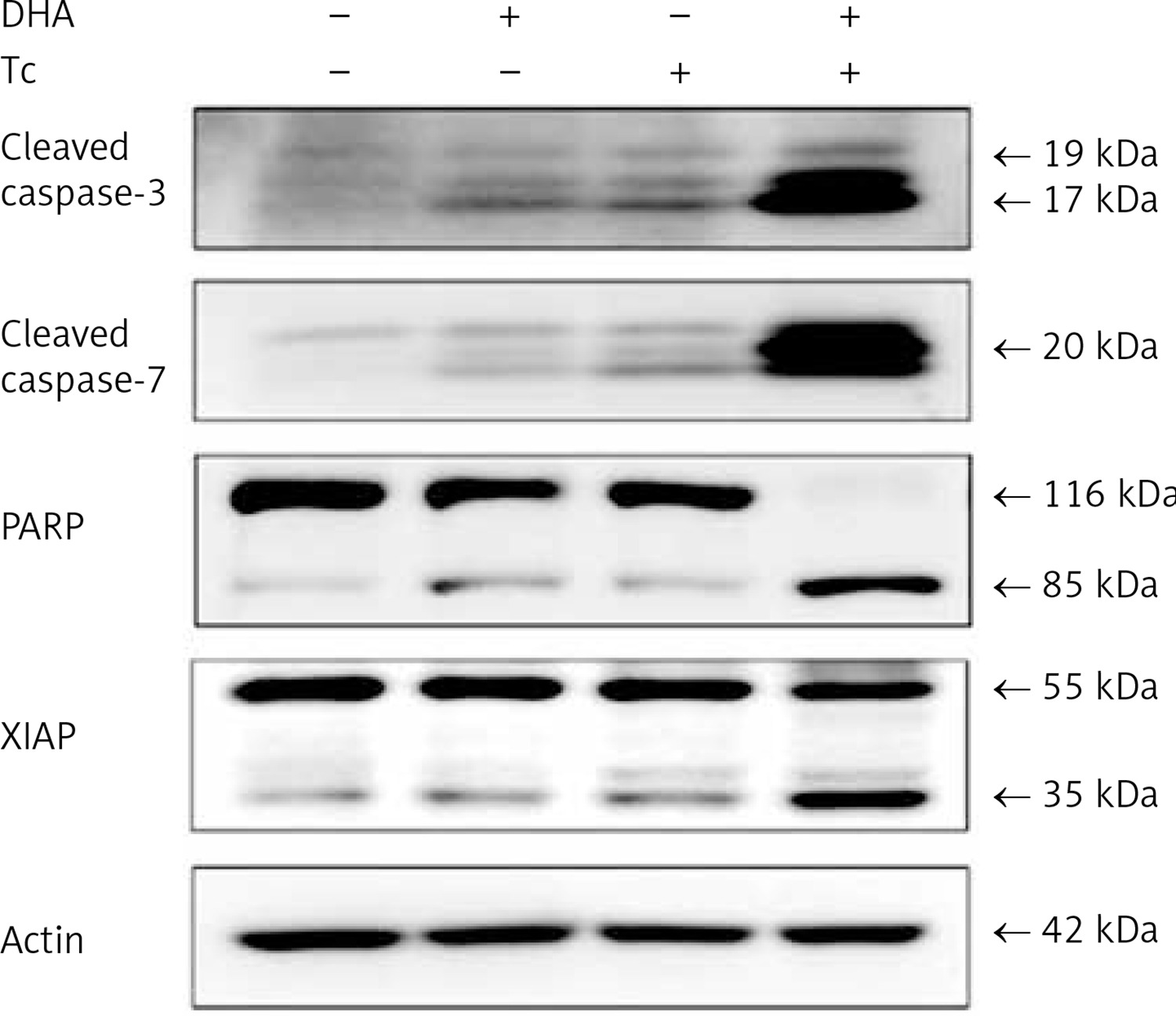
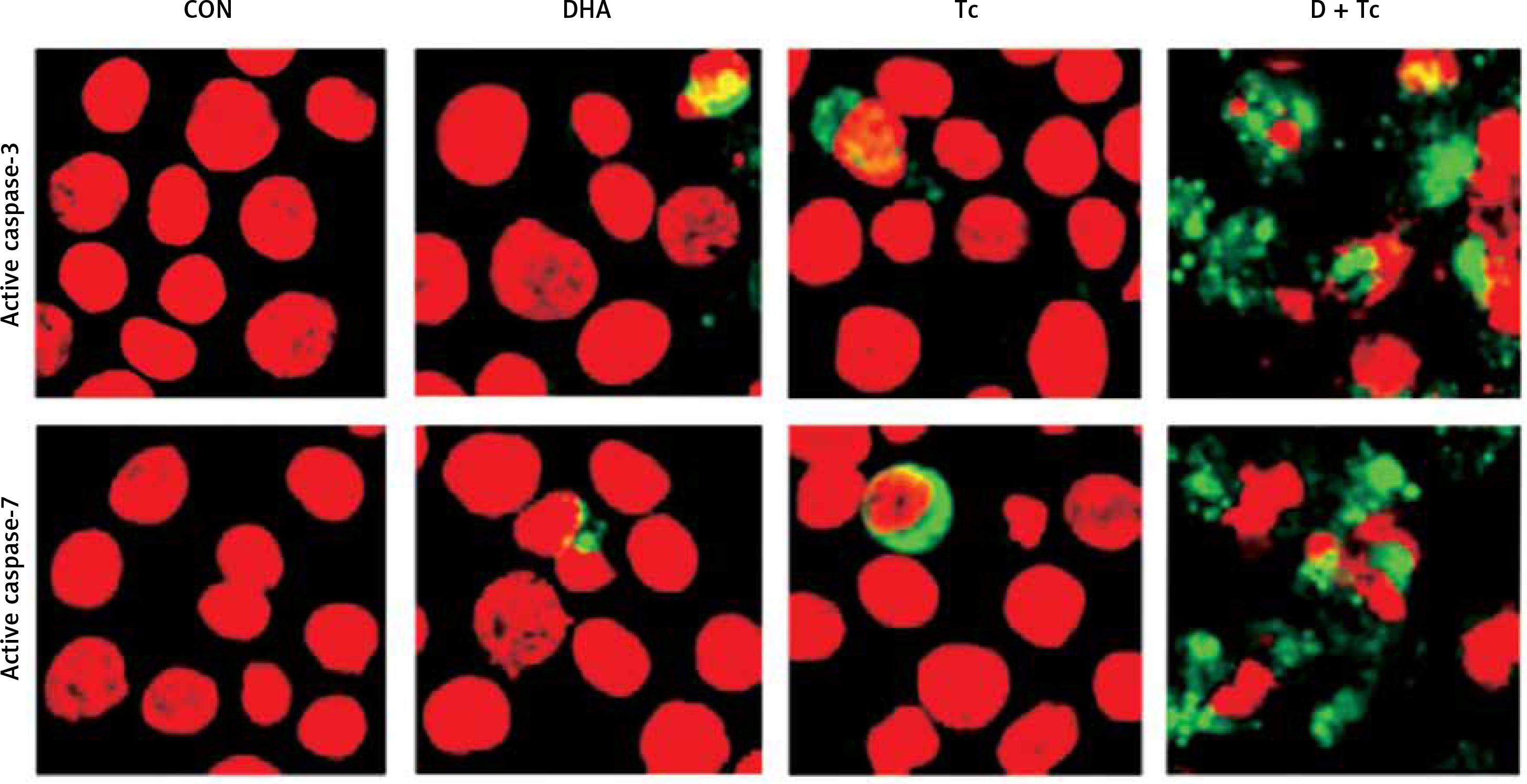
Figure 5
Immunocytochemical localisation of active caspase-3 and -7 in RL95-2 cells following exposure to combined treatment with DHA and triacsin C. Cells were treated with 125 μM DHA, 5 μM triacsin C, or combined treatment with 125 μM DHA and 5 μM triacsin C for 24 h, cytospun, fixed, and immunostained with a cleaved caspase-3 or -7 antibody, respectively. Propidium iodide (PI, red) was employed for differential nuclear staining
Microphotographs were taken using confocal microscopy. Original magnification: 800×.
Next, to explore the mechanisms by which DHA and triacsin C induce the extrinsic and intrinsic apoptosis pathways, activation of caspase-8 and -9 and BID was examined. Western blot analysis showed that the levels of cleaved caspase-8 (43/41 and 18 kDa) and -9 (37/35 kDa), and tBID (17 kDa), increased significantly with the combined treatment compared to DHA or triacsin C alone (Figure 6).
Figure 6
Activation of caspase-8, BID, and caspase-9 in RL95-2 endometrial cancer cells following exposure to combined treatment with DHA and triacsin C, as shown by western blotting analyses for procaspase-8, cleaved caspase-8, proBID, truncated BID (tBID), procaspase-9, and cleaved caspase-9. Cells were treated with 125 μM DHA, 5 μM triacsin C, or combined treatment with 125 μM DHA and 5 μM triacsin C for 24 h. Equal amounts of protein (30 μg) were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and immunoblotted using procaspase-8, cleaved caspase-8, proBID, tBID, procaspase-9, and cleaved caspase-9 antibodies, respectively. Actin was used as an internal (loading) control

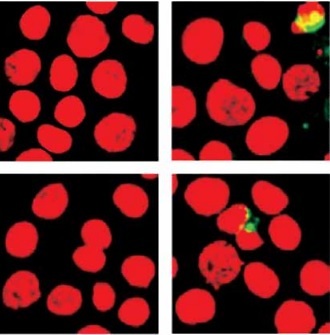
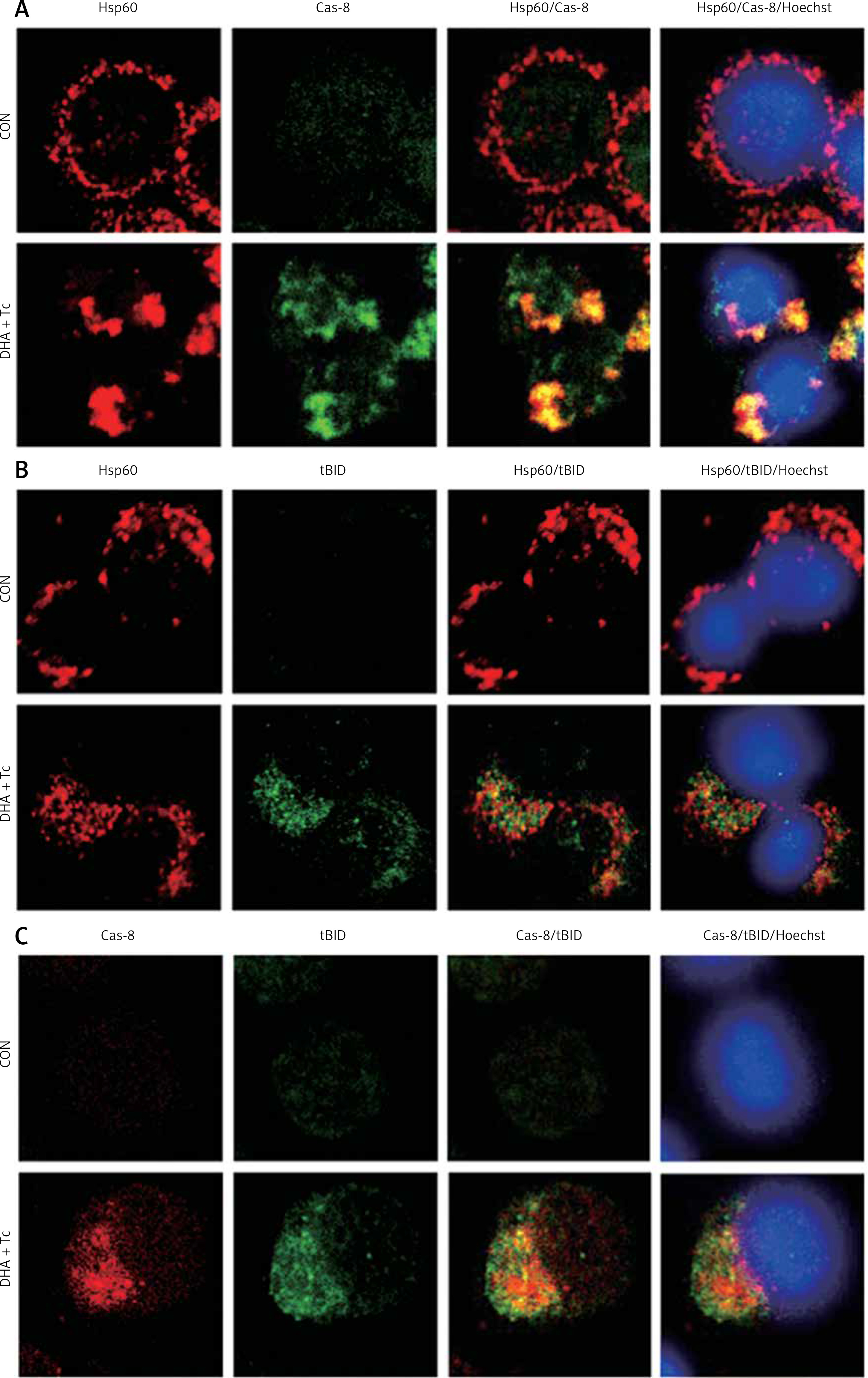
Activated caspase-8 directly activates caspase-3 or cleaves a BH3-only member of the Bcl-2 family of proteins, triggering its translocation to the mitochondria, where it causes release of cytochrome c. To explore the contribution of caspase-8/tBID to mitochondrial function, the subcellular localisation of active caspase-8 and tBID was confirmed by double immunostaining with active caspase-8 and tBID using mt-hsp60 as a mitochondrial marker protein. As shown in Figures 7 A and B, immunoreactivity of active caspase-8 and tBID can be mostly seen at the mitochondria following exposure to DHA and triacsin C. To examine whether caspase-8 co-localises with tBID at the mitochondria, double immunostaining was conducted with confocal analysis. The fluorescence signal of the active portion of caspase-8 and tBID produced a yellow image when superimposed (Figure 7 C), suggesting that the 2 molecules were co-localised in the mitochondria. Our results suggest that combined treatment with DHA and triacsin C induces caspase-8 translocation to the mitochondria, where it binds BID, leading to activation of tBID in the mitochondria.
Figure 7
Immunocytochemical localisation of active caspase-8 in RL95-2 cells following exposure to combined treatment with DHA and triacsin C. Cells were treated with 125 μM DHA, 5 μM triacsin C, or combined treatment with 125 μM DHA and 5 μM triacsin C for 24 h, cytospun, fixed, and immunostained with cleaved caspase-8 (A), tBID (B), cleaved caspase-8, tBID (C), and Hsp60 (as a specific mitochondrial protein marker) antibodies, respectively. Hoechst 33258 (blue) dye was employed for differential nuclear staining. Co-localisation of active caspase-8 (green) and Hsp60 (red) on mitochondria following exposure to combined treatment with 125 μM DHA and 5 μM triacsin C for 24 h
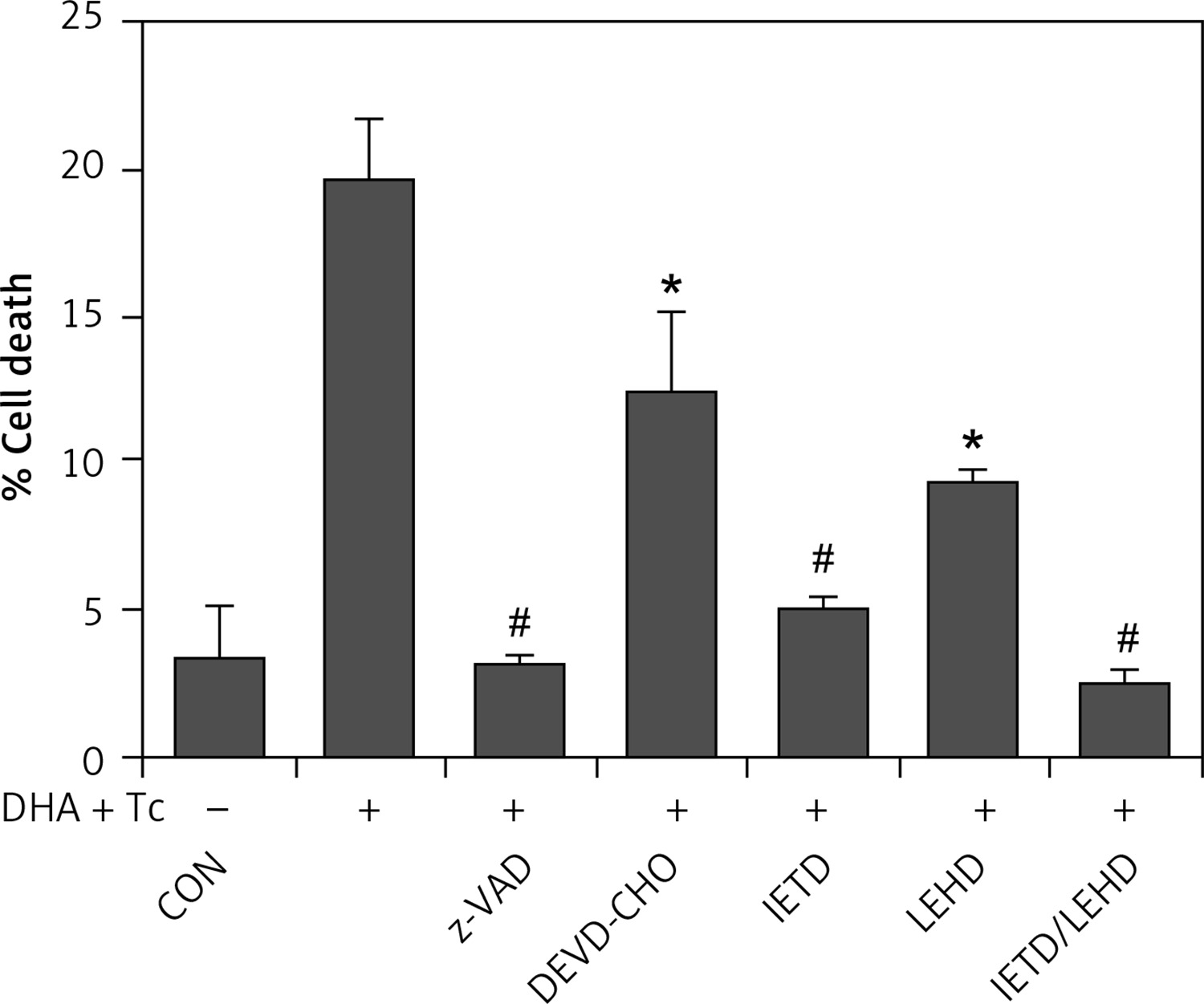
The requirement of caspase activation for DHA and triacsin C-induced apoptosis was examined using the following caspase-specific inhibitors: zVAD-FMK (pan-caspase inhibitor), DEVE-CHO (caspase-3/-7 inhibitor), IETD-CHO (caspase-8 inhibitor), and LEHD-CHO (caspase-9 inhibitor). RL95-2 cells were pretreated for 2 h with the inhibitors prior to treatment for 14 h with DHA and triacsin C. As shown in Figure 8, the pan-caspase inhibitor (zVAD-FMK), caspase-8 inhibitor (IETD-CHO), and a combination of caspase-8/-9 inhibitors (IETD-CHO/LEHD-CHO) completely attenuated DHA and triacsin C-induced cleavage of caspase-8, -9, and -7, and PARP. However, the caspase-3/7 (DEVD-CHO) and caspase-9 inhibitor (LEHD-CHO) partially blocked cleavage of caspase-3, -9, and -7, and PARP, but not caspase-8. The p17 fragment of caspase-3 was only weakly detected following pre-incubation with the pan-caspase inhibitor and caspase-8 and -8/9 inhibitors during combined treatment. Sub-G1 analysis showed that DHA and triacsin C-induced apoptosis was partially suppressed by the caspase-9 and -3/7 inhibitors, whereas it was completely blocked by the pan-caspase, caspase-8, and caspase-8/9 inhibitors (Figure 9). These results indicate that DHA and triacsin C-induced apoptosis is triggered through activation of caspase-8 and subsequent activation of caspases-9, -3, and -7.
Figure 8
Suppressed activation of caspase-8, -9, -3, and -7 by caspase inhibitors in RL95-2 endometrial cancer cells following combined treatment with DHA and triacsin C. Cells were exposed to 125 μM DHA and 5 μM triacsin C for 14 h in the absence or presence of zVAD-FMK (pan caspase inhibitor), DEVE-CHO (caspase-3/-7 inhibitor), IETD-CHO (caspase-8 inhibitor), LEHD-CHO (caspase-9 inhibitor), or IETD plus LEHD, respectively. Changes in caspases by caspase inhibitors in RL95-2 cells following exposure to DHA and triacsin C are shown by western blot analyses for caspase-8, -9, -3, and -7, and PARP cleavage. Equal amounts of protein (30 μg) were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and immunoblotted using a cleaved form-specific antibody for caspase-8, -9 -3, and -7, and PARP antibody. Actin expression was examined as a loading control
“-” – DHA and triacsin C without treatment, “+” – 125 μM DHA and 5 μM triacsin C treatment for 14 h.
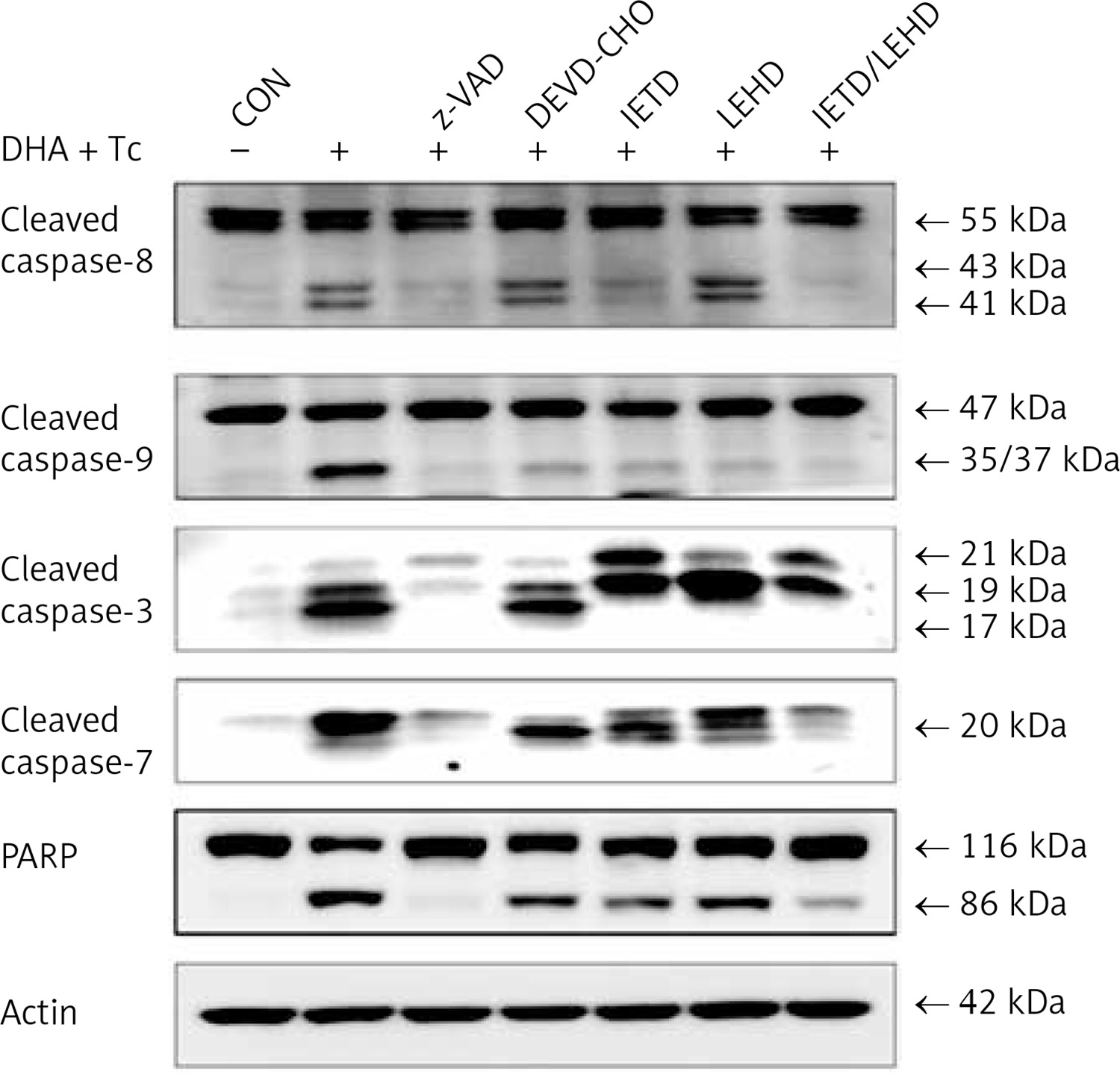
Figure 9
Suppression of apoptosis induced by combined treatment with DHA and triacsin C in the presence of caspase inhibitors in RL95-2 endometrial cancer cells. Cells were exposed to 125 μM DHA and 5 μM triacsin C for 14 h in the absence or presence of zVAD-FMK (pan caspase inhibitor), DEVE-CHO (caspase-3/-7 inhibitor), IETD-CHO (caspase-8 inhibitor), LEHD-CHO (caspase-9 inhibitor), or IETD plus LEHD, respectively. Cell death was assessed by flow cytometry. The percentage of cells with a sub-G1 DNA content was taken as a measure of cell death
At least 3 independent experiments were performed, and data shown are mean ± standard deviation. *,#p < 0.05 and p < 0.001 compared with the DHA and triacsin C-treated group, respectively. “-” – DHA and triacsin C without treatment, “+” – 125 μM DHA and 5 μM triacsin C treatment for 14 h.
Discussion
In recent decades, DHA has emerged as a promising anticancer agent. Although it exerts a selective cytotoxic effect on malignant tumour cells at physiological concentrations [9, 10], DHA alone was insufficient in the present study to induce apoptosis in the RL95-2 endometrial carcinoma line. ACS family enzymes, which activate and convert long-chain fatty acids, play a dominant role in phospholipid biosynthesis, β-oxidation, and lipid metabolism [24, 25]. Therefore, we hypothesised that inhibiting ACS function would disrupt DHA metabolism, leading to the accumulation of exogenous DHA in the mitochondria and contributing to the induction of apoptosis in RL95-2 endometrial cancer cells. This study showed that 125 μM DHA or 5 μM triacsin C alone did not induce apparent cytotoxic effects. However, combined treatment with 125 μM DHA and 5 μM triacsin C increased the sub-G1 population and apoptotic fragments. Western blotting also demonstrated that DHA and triacsin C induced apoptosis through the mitochondrial pathways via caspase-9, -3, and -7, as well as through the extrinsic pathway by activating caspase-8/BID.
Mitochondria are particularly susceptible to oxidative stress; several studies have demonstrated that the accumulation and association of DHA with cardiolipin may lead to the generation of ROS and changes in cardiolipin and apoptosis signalling [31–33]. In addition, inhibiting ACS with triacsin C leads to the accumulation of exogenous free fatty acids [34, 35], which might affect mitochondrial status in cells undergoing apoptosis. Cardiolipin is generally believed to be localised at the inner membranes of mitochondria, but is also present at the contact sites formed between mitochondrial inner and outer membranes [36, 37]. Cardiolipin has also been shown to act as an activation platform for bringing together both caspase-8 and tBID. During apoptosis, the translocation of caspase-8/tBID at contact sites between the inner and outer mitochondrial membranes leads to a loss of cardiolipin content and dissociation of cytochrome c from the mitochondrial inner membrane [36, 38–40].
Therefore, it has been postulated that inhibition of ACSs by triacsin C would induce activation of caspase-8/tBID in RL95-2 cells by dissociating cytochrome c at the mitochondria membrane. The activation of caspase-8/tBID and subsequent translocation to the mitochondrial membrane (dependent on cardiolipin) is critical for inducing apoptosis and may affect mitochondrial membrane function and integrity, sequentially leading to ROS production in the electron transport chain [41]. In addition, inhibiting ACSs with triacsin C modifies the structure and content of cardiolipin [42], suggesting that ACSs are essential for the maintenance of cardiolipin content as well as cell survival.
Some limitations in our study should be mentioned. We did not observe the effect of apoptosis in heterogeneous endometrial cells treated with triacsin C and DHA. Therefore, it will be necessary to evaluate the combined effect on endometrial cancer samples, such as heterogeneous tissue, in a further study. However, this is the first study to verify the apoptotic effect of the combination on endometrial cancer cells. The mechanisms of DHA and triacsin C-induced apoptosis may provide the basis for developing therapeutic strategies for endometrial cancer.
In conclusion, the synergistic effect between DHA and triacsin C has not been investigated in most clinical and preclinical studies. Nevertheless, further elucidation of the apoptotic mechanisms involving DHA and ACS inhibition could shed light on possible new treatment strategies for endometrial cancer.