Introduction
Obesity prevalence has doubled in more than 70 countries and continuously increases globally since 1980 [1]. Studies have associated high body mass index (BMI) and physical inactivity with a set of chronic diseases such as type 2 diabetes (T2DM), and an array of other disorders [2–4]. The main link between these metabolic disorders is the ability to induce insulin resistance and, as a consequence, affect the whole organism’s function. However, some organs and tissues exacerbate the pathological conditions including: 1) adipose tissue (AT) – the site of fat accumulation, 2) the gut – the site for the microbiota and metabolites that have been associated with metabolic disorders [5], 3) muscles – the primary site of insulin resistance [6], 4) the liver – obesity is a major risk factor for liver damage, and finally, 5) the pancreas – once impaired it leads to compromised insulin production and secretion. All metabolic processes that these organs are involved in are also influenced by immunological responses that stimulate and maintain them.
The interface between the immune system and metabolism has been investigated over the last 15 years and has been branded with the term immunometabolism. This interdisciplinary approach made the field essential for understanding the pathology and progression of metabolic diseases as immunometabolism places the low-grade chronic inflammation as the central cause and consequence of metabolic disorders [7]. Inflammation is described as a prompt and a short-term response to deal with injuries and infections, providing repair to injured tissues, and it is composed of a series of signals and pathways that are rapidly resolved upon healing. In contrast, low-grade chronic systemic inflammation or metaflammation is primarily caused by persistent activation of the innate immune system that promotes increased production and secretion of proinflammatory cytokines and other mediators [8, 9]. It is generally believed that persistent over-nutrition, physical inactivity and exposure to certain epigenetic factors contribute to the development of low-grade systemic inflammation associated with metabolic diseases [10–12]. The constant activation of the innate immune system has been shown to induce the production of stimuli that may additionally activate the adaptive immune system. In some tissues, such as visceral AT, an alternative chain of events combining the immune response and inflammation was described, in which the adaptive immune cells (CD4 and/or CD8 T cells) were shown to trigger AT inflammation [13, 14]. Together, it is proposed that an interplay between disturbed metabolic state and these low-grade chronic inflammatory responses culminate in a vicious cycle leading to the development of metabolic diseases, such as T2DM [15–17].
An inflammatory state playing a role in the development of metabolic diseases was shown for the first time in 1993 [18] when the adipose tissue (AT) was described to produce the proinflammatory cytokine tumor necrosis factor α (TNF-α). In accordance, it was proposed that obesity could be associated with enhanced expression of proinflammatory mediators and that this environment could modulate glucose metabolism and/or insulin action [19].
Increased serum free fatty acids (FFAs) levels have been associated with insulin resistance in obese individuals [20–23]. Especially saturated FFAs have been correlated with induction of the inflammatory response and insulin resistance in insulin target tissues, while polyunsaturated FFAs have been described as generally anti-inflammatory [24]. In contrast to omega-6 FFAs, omega-3 FFAs by stimulating the biosynthesis of specialized pro-resolving lipid mediators (SPMs; such as protectins, resolvins, lipoxins, maresins) in immune cells and other tissues are believed to possess a strong protective anti-inflammatory potential [25]. Specialized pro-resolving lipid mediators were shown to improve insulin sensitivity and reduce AT inflammation via inhibition of TNF-α, IL-1β, IL-6 and IL-8 secretion [26].
The analysis of AT from obese patients showed that macrophages were able to infiltrate this tissue [27] and that FFAs promoted the polarization of these cells towards a proinflammatory phenotype (M1 macrophages) [28]. It is important to mention that macrophage polarization has been clustered into two major macrophage polarization programs, classically activated macrophages or M1 and alternatively activated macrophages or M2, each related to specific immune responses, among which both progression and resolution of inflammation constitute critical determinants [29]. However, this clear distinction has been challenged with data identifying a metabolically activated macrophage phenotype that is mechanistically distinct from M1 or M2 activation [30, 31].
Nevertheless, the presence of classical M1 macrophages in AT of obese patients and high-fat fed animal models (HFD; HFD-fed M-JAK2–/– and HFD-fed MIF–/– C57Bl\6J) was clearly associated with impaired insulin action [32, 33]. Beyond the innate immune system, it has also been demonstrated that the adaptive immune response with T and B lymphocytes may influence metabolic processes. So far, the immuno-metabolic crosstalk has been described in various tissues, suggesting functional links with consequences for translational studies [34, 35].
This review aims to present and discuss the updated knowledge about important processes in the intercommunication between the immune system and metabolism (Figure 1). Although much of what is known about these interactions during obesity and obesity-related diseases was first described in AT, other organs are also involved and they will be discussed in more detail in forthcoming sections.
Figure 1
Important processes in the intercommunication between the immune system and metabolism
[This figure was created using images from Servier Medical Art Commons Attribution 3.0 Unported License. (http://smart.servier.com). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License]
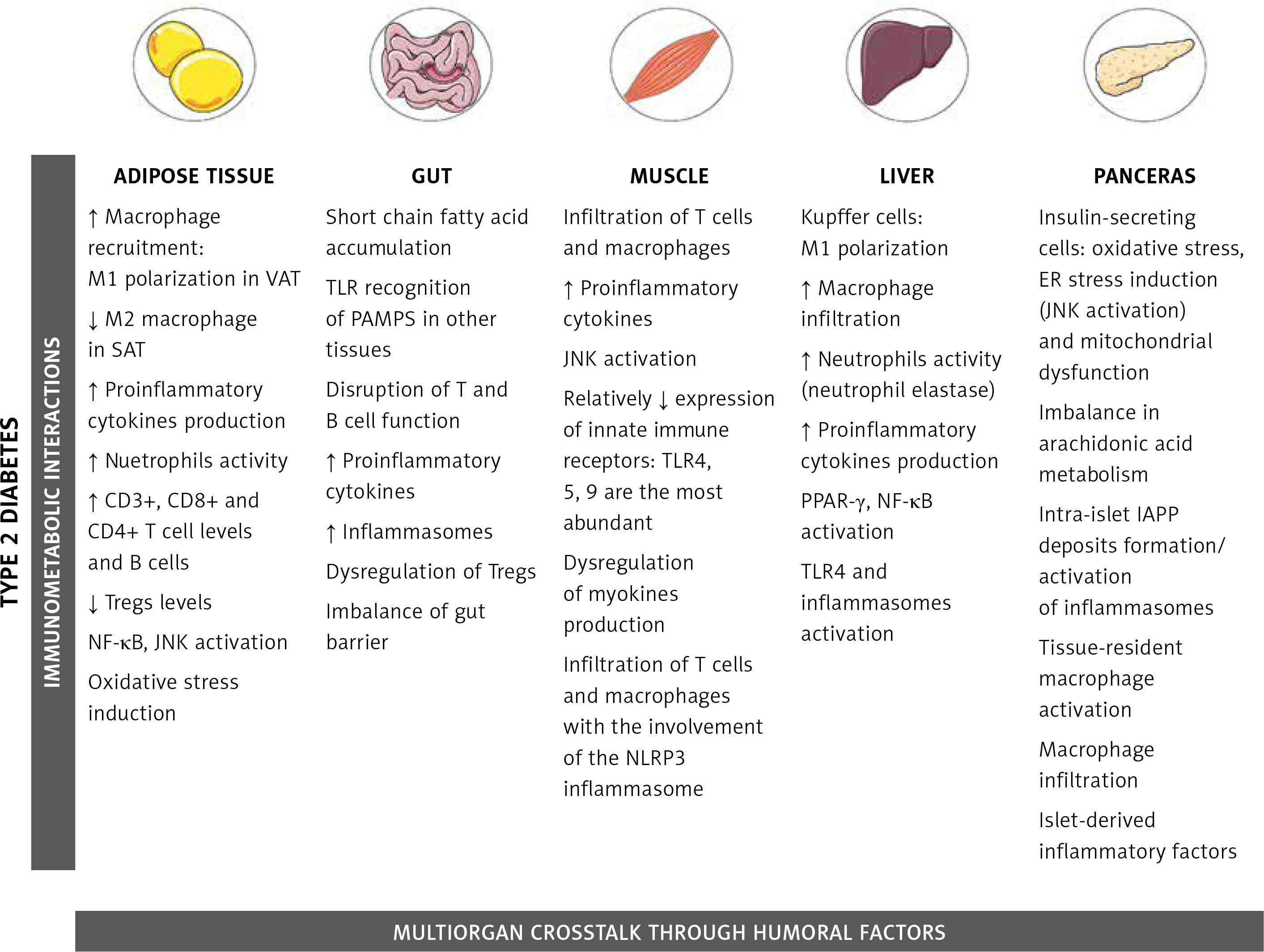
Tissue-specific immune responses leading to metabolic diseases
Adipose tissue
Initially treated as a deposit for triacylglycerol and thus as a sole energy-storage tissue, the AT is now considered a multifunctional endocrine organ that is able to synthesize bioactive factors (adipokines) to regulate metabolism, energy intake, fat storage and immunity [36, 37]. Composed mainly of adipocytes, the AT also contains pre-adipocytes, endothelial cells, fibroblasts, and a diversity of immune cells such as macrophages, neutrophils, T lymphocytes, and others, with clear differences observed between obese and lean adipose tissue, as well as distinct functions of visceral fat (VAT) and subcutaneous fat (SAT) [38, 39]. The volume of abdominal visceral fat area was the most predictive factor for AT macrophage infiltration in patients [39], and correlated with increased proinflammatory mediator secretion [40]. In lean individuals only 10% of AT is composed of macrophages and they are predominantly in the anti-inflammatory M2 state [41]. In contrast, in obese individuals up to 50% of AT consists of M1 macrophages [42, 43]. TNF-α released from M1 macrophages can inhibit the transcription factor PPARγ that is responsible for the ability of AT to produce new healthy fat cells from stem cells [44]. Consequently, the decreased capability for generation of new healthy fat cells together with a parallel overexpansion of inflamed adipocytes results in the acceleration of necrotic cell death of adipocytes. This process triggers the aggravation of AT inflammation through migration of neutrophils and macrophages [45–47].
Alterations in the AT immune status influence cytokine content, adipocyte metabolism and insulin sensitivity. Proinflammatory adipokines stimulate local recruitment and accumulation of inflammatory cells in AT as well as increasing the systemic levels of inflammatory markers [48–50]. This is the mechanism suggested to trigger the low-grade chronic inflammation that is directly related to the development of various diseases, such as obesity, T2DM, cardiovascular pathologies or cancer [51, 52].
The major immuno-metabolic interaction taking place in the obese AT is the adipocyte-macrophage crosstalk [53]. The inflamed environment is not only a result of the TNF-α production by adipocytes [18] but also a result of macrophage activity. M1 macrophages secrete high levels of chemokines and proinflammatory cytokines, fostering the insulin-resistant state in AT [16, 54]. It has been shown that amelioration of AT inflammation strongly correlates with a decreased number of proinflammatory macrophages as well as reduction of the whole-body insulin resistance [55, 56]. Other proinflammatory factors that impair insulin signaling in the AT include activation of the nuclear factor-κB (NF-κB) [57, 58], and c-Jun N-terminal kinase (JNK) [59] pathways in adipocytes, as well as induction of oxidative stress [60]. Once active, these components stimulate the transcription of genes that, 1) encode pro-inflammatory proteins, 2) inhibit the activation of the insulin receptor, and, 3) impair processes such as the PI3K/Akt/mTOR pathway resulting in defective insulin signaling [61].
Interleukin-6 (IL-6), secreted by the adipocytes, stromal cells and macrophages, also affects insulin sensitivity in the AT through similar mechanisms and its serum level positively correlates with the degree of obesity in humans [62–64]. Although its activity impairs insulin signaling [65], the absence of IL-6 leads to the development of obesity and insulin resistance [66, 67], suggesting that a certain threshold of IL-6 concentration is required for unbiased AT function. The appetite-control adipokine leptin is another factor considered as a crucial pro-inflammatory contributor to AT dysfunction [68], capable of activating macrophages [69], and promoting glucose metabolism in CD4+ Th1 cells [70–72]. Finally, neutrophils, which are the first immune cells recruited to the AT in obese animals fed a HFD [73], have also been shown to participate in the AT inflammatory response and malfunction. When present in the AT, neutrophils secrete neutrophil elastase, which hampers insulin signaling through degradation of the insulin receptor substrate 1 (IRS-1) [73]. This process was shown to be attenuated in HFD exercise-trained mice [74].
Interestingly, apart from the innate immune cell components, lymphocytes have also been shown to be engaged in the regulation of the inflammation-metabolic state axis of AT. After macrophages, CD3+ T cells are the largest population of immune cells present in the AT with even more abundant presence in response to increasing adiposity [75]. Besides CD3+, the levels of CD8+ and CD4+ T cells are also elevated during obesity [76–78], mostly in VAT [79, 80]. The increase in CD8+ T cells is suggested to precede and contribute to the accumulation of macrophages in the AT, and their depletion is associated with the decrease of M1 macrophages and insulin resistance improvement [76]. The CD4+ (Th1) increase is suggested to have a pathological role in obesity and obesity-induced insulin resistance. It was demonstrated that activated CD4+ T cells (CD4+CD44hiCD62Llo) accumulate in the visceral AT of obese mice and display features of cellular senescence [81]. In addition, the MHC class 2 induction in the obese AT activates CD4+ T cells, which triggers AT inflammation and insulin resistance [82]. γδ T cells, when in the white adipose tissue (WAT) and after long-term HFD, are present in high numbers and secrete high amounts of IL-17, a cytokine that regulates adipogenesis and glucose metabolism [83]. Animals lacking γδ T cells display reduced HFD-induced inflammation, while the presence of these cells positively contributes to WAT inflammation by regulating the macrophage populations present in the tissue [84]. It is important to point out that the dynamics of the immune system within VAT and SAT is remarkably different [85, 86].
On the other hand, Th2 cells are suggested to play a protective role against systemic inflammation and insulin resistance by producing type 2 cytokines (IL-4, IL-5, IL-13) and stimulating polarization of macrophages to M2 phenotypes. Production of Th2 cytokines, such as IL-10, was reported to occur in SAT, indicating an anti-inflammatory role of this fat depot [87–89]. CD4+ T cells once transferred into a diet-induced obesity animal model were shown to acquire a Th2 profile. The observation was associated with a reduction of body weight and insulin resistance improvement [88, 90]. An unbalanced ratio between Th1 and Th2 cells is strongly associated with systemic inflammation and insulin resistance [88]. T regulatory cells (Tregs), a cell type that generally inhibits the acceleration of inappropriate inflammatory processes, thereby maintaining insulin sensitivity [91], are also involved. Tregs display reduced levels during obesity [92], while non-obese AT is rich in Tregs [14]. It was demonstrated that obese mice with adipocytes lacking MHC class 2 and consequently displaying lower amounts of IFN-γ presented a higher number of Tregs, which led to reduced obesity-induced AT inflammation and insulin resistance [92]. In addition, imbalances between Tregs and Th17 cells, characterized by the production of proinflammatory cytokines such as IL-17A, IL-22, and IL-21 [93], caused by lipotoxicity were shown to contribute to obesity and T2DM progression [94]. Ablation of Tregs specifically in the AT of HFD-fed animals resulted in impaired insulin sensitivity [92].
B lymphocytes have been shown to accumulate before T cells in the AT in HFD models [95] and are recruited by the pro-inflammatory chemokines produced by the AT (CXCL10/CCL2/CCL5) [96], and through leukotriene LTB4 signaling [97]. A pathogenic role for B cells was reported, leading to enhancement of the AT insulin resistance [98]. B cell accumulation is associated with M1 macrophage polarization, activation of T cells (CD4+ and CD8+), and production of pathogenic IgG antibodies [98]. A distinct B cell subtype, called the B regulatory cell (Breg), was reported as a constitutive subset with an anti-inflammatory profile within AT. Bregs maintain tissue homeostasis, produce IL-10, and their function is impaired during obesity [99]. Bregs were shown to have a central contribution to the progression of obesity-induced inflammation, displaying reduced numbers in the obese AT [99, 100]. The same study showed a causal relationship between increased levels of Th1 cytokines and decreased frequency of Bregs [100]. Another anti-inflammatory agent influencing the AT metabolism is the cytokine IL-37. Transgenic mice expressing IL-37 were found to be protected against metabolic syndrome even when fed a HFD [101]. Moreover, HFD-fed mice treated with recombinant IL-37 displayed improved insulin sensitivity and obesity-induced inflammation [102].
Moreover, dietary FFA composition has been suggested to play an important role in insulin resistance and AT inflammation [26, 103, 104], with saturated FFAs promoting AT inflammation whereas omega-3 FFAs resolves the inflammatory response [105, 106].
The innate lymphoid cells (ILCs) are a lineage-negative subset of T cells (lacking the expression of surface markers that define other T cells) that act in response to the cytokines produced by surrounding macrophages, dendritic cells, and epithelial cells [107]. ILCs comprise five different subsets of immune cells: 1) natural killer (NK) cells, 2) ILC1s that produce interferon γ (IFN-γ), 3) ILC2s that produce IL-5 and IL-13, 4) lymphoid tissue inducer cells, and 5) ILC3s that produce IL-17 and IL-22 [107, 108]. These cells are suggested to regulate metabolism and to play a role in the development of obesity. NK cells and ILC1s are involved in the development of obesity-associated insulin resistance [109, 110], ILC2S are involved in the browning of the WAT and protection against obesity [111], ILC3S and lymphoid tissue inducer cells might be involved in the induction of obesity and obesity-associated insulin resistance due to lymphotoxin/IL-23/IL-22 activity [112].
Cell death and hypoxia also contribute to AT macrophage migration through the formation of “crown-like structures”, and the hypoxia hypothesis, respectively. Macrophages accumulate in the AT around adipocytes that are dead or in the process of dying, forming “crown-like structures” around the dead adipocytes [113]. These macrophages (M1 phenotype) produce a range of pro-inflammatory cytokines, such as TNF-α, which ultimately results in the development of metabolic disorders [114]. Moreover, macrophages localized in the “crown-like structures” in obese AT were shown to be enriched in Mincle (macrophage-inducible C-type lectin) [115]. The expression of Mincle correlated with the intensity of AT inflammation and ectopic lipid accumulation [116].
Their proliferation has IL-4/STAT6 as the driving force since IL-4 administration significantly enhanced the proliferation of ATMs in non-obese animals [117]. However, AT macrophages were recently suggested to be responsible for promoting the clearance of dead adipocytes through lysosomal exocytosis, also indicating their beneficial role [31]. Finally, the hypoxia hypothesis states that during obesity, angiogenesis is insufficient to maintain the vascularization and oxygenation necessary for AT proper function. Hypoxia activates the hypoxia-inducible factors (HIFs) which can stimulate gene expression of proinflammatory pathway genes such as NK-κB [118], affect AT macrophage polarization and inhibit preadipocyte differentiation [119]. The understanding of obesity and insulin resistance development from the AT perspective can be summarized through the interactions between adipokines, immune cells, cell death, and hypoxia.
Gut
The gut is a site of intricate immunological processes since it is the largest site of contact with antigens either from microbiota or from dietary factors. It also possesses the largest mass of lymphoid tissue in the organism. In the past decade, the number of investigations about immuno-metabolic interactions within the gut increased exponentially, especially due to strong evidence suggesting a direct association of the gut microbiota composition and its metabolites with the development of obesity and related metabolic disorders [120, 121].
Data show that caloric restriction and obesity affect gut permeability [122–124]. Standardized caloric restriction positively impacted gut permeability through a mechanism that remains unclear [122]. On the other hand, intestinal barrier impairment was shown to be exacerbated by a lipid challenge in obese patients [123], and anthropometric measurements and metabolic variables were shown to be positively correlated to increase in gut permeability during obesity [124]. The metabolites and byproducts generated by the microbiota also play an important role as components influencing inflammatory and metabolic processes as well as modulating the intestinal barrier function [125–127]. Molecules such as acetate, propionate, and butyrate – short-chain fatty acids (SCFAs) – produced as a result of the fermentation processes performed by the microbiota, can act as signaling and regulatory molecules involved in inflammation and insulin sensitivity [128–131]. Under non-obese conditions, SCFAs do not accumulate since they are transported through the portal vein, reaching the liver for clearance [132]. However, during obesity, the outcome of the increased barrier permeability is migration of products that usually remain in the intestinal environment, but which are now directed towards the systemic circulation at high concentrations. The problem associated with this migration is the recognition by the immune system of pathogen-associated molecular patterns (PAMPS), and lipopolysaccharides (LPS) in other tissues [133]. This recognition through toll-like receptors (TLRs) stimulates the proinflammatory response in insulin target tissues, contributing to reduced insulin sensitivity [134, 135]. On the one hand, excessive SCFAs serve as an additional source of energy as well as an inflammatory factor in tissues such as the AT and liver. At the same time, they are involved in the β-cell glucose-stimulated insulin secretion through the G-protein coupled receptor 43 (GPR43)/GRPR41 [136, 137], and release of pancreatic peptide YY3-36 and glucagon-like peptide-1 (GLP-1) [138]. This suggests that the composition of the microbiota is responsible for the generation of a different type of SCFAs, which in turn are capable of triggering different regulatory cascades.
Germ-free animals enable greater insights to be gained into the impact of the microbiome in the metabolic homeostasis. These animals are resistant to the development of obesity and insulin resistance [139], concomitantly to the disruption of T and B cell function, and less efficient, impaired Tregs [140]. Obesity leads to dysbiosis [141], and it was recently suggested that this imbalance occurs even in a diet-independent fashion [142]. HFD affects not only the gut but also the gastric microbiota [143], and germ-free mice that are long-term exposed to a microbiota-derived from HFD animals develop dysglycemia and glucose intolerance [144]. An array of studies indicate that the diversity of the microbiota is closely associated with disease development and show that reduced diversity is positively correlated with inflammation and insulin resistance [145, 146]. For this reason, many efforts have been made to identify the potential differences between microbiota in health and disease [147].
Obesity stimulates the accumulation of non-beneficial bacterial strains, the conclusion made by experimental transfer of microbiota from obese to germ-free mice resulting in increased adiposity [148]. As a consequence of this imbalance, PAMPs and LPS stimulate a pro-inflammatory environment within the gut. High fat or high sugar diets were shown to induce imbalance in the ratio of specific strains of bacteria within the gut microbiota (Firmicutes/Bacteroidetes) and increase amounts of pro-inflammatory strains such as Proteobacteria [149, 150]. These alterations were partially restored by reverting to a regular chow diet [151]. Changes in the Firmicutes/Bacteroidetes ratio are associated not only with obesity (high F/B), but also with weight loss (low F/B) [152]. Obese and lean humans were found to display comparable altered taxonomic features [153]. Lactobacillus spp. are also affected. Rats that went through short- and long-term periods of caloric restriction displayed increased proliferation of this genus [154]. Lactobacilli are probiotics with mainly anti-inflammatory effects [155] capable of regulating Th17/Treg differentiation [156], altering the Th1/Th2 ratio [157], and suppressing macrophage WAT infiltration [158]. Under physiological conditions, microbe-associated molecular patterns (MAMPS) stimulate the production of anti-inflammatory factors promoting tolerance and proper function of the intestinal barrier [159, 160]. On the other hand, during obesity (diet-induced), where the intestinal barrier is known to be more permeable [161], MAMPS stimulate intestinal epithelial cells, macrophages, and dendritic cells to produce pro-inflammatory cytokines [162]. The activation of inflammasomes is also suggested to contribute to gut microbiome perturbations [163, 164]. However, a recent rigorous microbial phylogenetic analysis performed in inflammasome-deficient mice failed to reproduce the gut microbiota composition alterations, raising the importance of careful experimental procedures and controls in evaluating results about the gut microbiota [165].
Over the last decade, the investigation of the interaction between the gut metabolism and the immune system has expanded our understanding about its impact on health and disease. Biomarkers to discriminate specific microbes’ species will soon confirm or refute the direct role of bacterial strains in obesity, T2DM, metabolic disorders and cancers [166, 167]. Although the exact mechanisms are still not fully understood, dysbiosis has an impact on microbe and host metabolism, as well as shaping inflammatory responses. The complex crosstalk between the microbiota, intestinal permeability and inflammation that leads to insulin resistance, alterations in the glucose metabolism, and T2DM has already been reviewed by different authors [168–170]. Evidence accumulated so far opens the field for the future development of therapeutic strategies.
Skeletal muscle
The skeletal muscle (SM) is the primary site for dietary glucose uptake and storage in the form of glycogen, being, consequently, a crucial component affected during the development of insulin resistance [6]. The physiology behind how the SM takes up glucose is extensively investigated, with special attention to the insulin signaling cascade and glucose transporter 4 (GLUT4) translocation regulation [171, 172]. However, very little is known about the potential role of the immune system in this regulatory mechanism or how inflammation impacts muscle metabolism.
The very first link reporting immuno-metabolic interactions influencing muscle physiology was the observation that LPS, when injected into dogs, leads to insulin resistance caused by impairment of SM glucose uptake [173]. Years after, it was shown that, like the AT, the skeletal muscle of obese animals and humans can also generate TNF-α [174], and its attenuation was associated with improved insulin sensitivity and glucose metabolism [175]. The JNK pathway is also involved, and its role in the pathogenesis of obesity-induced insulin resistance is well described [176, 177]. The mechanisms behind the protective effect of global JNK deficiency against diet-induced insulin resistance were carefully discussed previously [176]. While some studies have shown the SM-specific ablation of JNK results in an improvement of insulin resistance an improvement of insulin resistance [178–180], others indicate no impact [181, 182]. Thus, this immuno-metabolic interaction is still under discussion.
Similarly to the AT, many of the classical innate immune components play a role in the SM metabolism. The SM is characterized by a relatively low expression level of innate immune receptors [183]. Of all innate immune receptors, TLR4, 5, and 9 are the most abundant [184]. TLR4 activation stimulates glycolysis, inhibits fatty acid oxidation and induces insulin resistance [185]. Pharmacological inhibition of this receptor was shown to protect mice against diet-induced obesity [186]. While the whole-body deficiency of TLR5 causes increased fat mass, insulin resistance and metabolic syndrome-like features [187], the TLR5 SM-specific contribution remains unclear so far. The most recent update explains the role of TLR5 in smooth muscle and the development of atherosclerosis through activation of TLR5-dependent NADPH oxidases, and H2O2 generation [188]. TLR9, in turn, has been suggested to be involved in the development of type 1 diabetes [189], and despite being the most abundant TLR at the mRNA level in muscle, its role in SM metabolism is being investigated. The role of other TLRs has been extensively reviewed [183].
Although often neglected as a secretory tissue, myocytes can express and secrete myokines. The subset includes some cytokines (IL-6, IL-8, IL-15), fibroblast growth factor 21 (FGF21), basic FGF (FGF2), follistatin-related protein 1 (FSTL-1) and other molecules [190]. The myokine activity counterbalances the effects of the adipokines, stimulating beneficial effects on glucose and lipid metabolism and inflammation [190–192]. The SM-derived IL-6 is the most investigated myokine and, besides controversies [192], it is suggested to contribute to the metabolic homeostasis reestablishment upon exercise but not under basal conditions [193]. Moreover, IL-6 was reported to act in a gender-specific manner [194]. Mitochondrial dysfunction [195] and ER stress [196] trigger FGF21 secretion, but the relationship between FGF21-mediated metabolic alterations and disease progression is still not clear [197].
Altogether, the secretion of myokines does not seem to be the factor responsible for the development of muscular inflammation during obesity. Unlike in the AT, it is suggested that the inflammation in the muscles develops as a result of the production of proinflammatory molecules (adipokines) secreted from accumulated intermuscular and perimuscular fat depots and not by the tissue itself [198]. The obesity-induced increase of such fat storage sites is correlated with the development of a pro-inflammatory environment in the muscle [198], influencing insulin sensitivity by impairing its signaling as well as glucose uptake through the GLUT4 reduction [199].
Apparently, the skeletal muscle is more of a target of the inflammation induced by insulin resistance in other organs than, in fact, a site where this inflammation begins. The most accepted hypothesis is that free fatty acids (FFAs) stimulate the inflammatory response characterized by infiltration of T cells and macrophages with the involvement of the NLRP3 inflammasome [198, 200–202]. Likewise, it occurs in other tissues; macrophages in the SM polarize towards a proinflammatory phenotype during obesity [203]. Consequently, proinflammatory mediators such as TNFα, IFN-γ, and IL-β are shown to be augmented, while anti-inflammatory markers, such as IL-10, remain unaffected [204]. Similarly to AT, omega-3 FFAs were shown to restore SM insulin sensitivity and ameliorate lipotoxicity [205].
In summary, despite the muscle’s ability to secrete myokines, the majority of the inflammatory molecules affecting its metabolism originate from the so-called perimuscular AT and not from the muscle itself. In the context of the immuno-metabolic interactions, the impact of SM has been under-investigated and the role of this communication and its implications for the development of metabolic diseases are still largely unknown.
Liver
The liver plays a crucial role in detoxification of xenobiotics, protein synthesis, carbohydrate household, lipid and protein metabolism, iron homeostasis, and secretion of hormones (IGF-1 and hepcidin). It is also known that the hepatic tissue is immunologically complex, being responsible for the production of cytokines, chemokines, and complement components, containing a diverse population of immune cells [206]. The hepatic immune system is regularly challenged with dietary factors of high inflammatory potential. The combination of constant metabolic activity and regular exposure to proinflammatory factors contributes to the state of chronic low-level inflammation of this organ [12]. Disruptions of this close immuno-metabolic interaction are associated with pathological inflammation that can lead to liver fibrosis, cancer, non-alcoholic fatty liver disease (NAFLD), obesity and other chronic diseases [207–210].
The liver displays two dominant types of macrophages: the Kupffer cells (KC) and the monocyte-derived macrophages [211]. Kupffer cells are liver-resident macrophages, comprising up to 90% of the total population of macrophages in the organism and around 25% of the whole subset of non-parenchymal cells in the organ [212]. In contrast to other macrophages, KC are prone to respond in a milder manner and are known to be able to secrete high concentrations of IL-10 [213]. Metabolic disorders do not impact KC in a quantitative way but impact their polarization states [214]. Under normal conditions, due to the tolerance required in the hepatic environment, KC tend to exhibit an M2-like phenotype [214]. On the other hand, because obesity and hepatic steatosis stimulate 1) secretion of higher levels of TGF-β and other proinflammatory cytokines [214], and 2) interaction between PPARγ and NF-κB [215] signaling pathways, KC are polarized towards a proinflammatory phenotype (M1). The imbalance in the M1/M2 ratio was shown to be restored in the HFD-induced NAFLD animal model upon treatment with rosiglitazone, a thiazolidinedione [216]. It indicates that PPARγ modulation affects the interaction with NF-κB, reducing M1 polarization and ameliorating hepatic steatosis [216]. It is suggested that TNF-α and IL-1β are crucial players in the development of NAFLD and KC are the main generators of the first one in a process mediated by TLRs [217]. IL-1β meanwhile was shown to be important for the progression of NAFLD to NASH [218].
Phenotypically different from KC is the other important group of macrophages found in the liver, the macrophages that are recruited to the organ – monocytes-derived macrophages [219]. These macrophages originate from blood monocytes and have CLEC5A as a specific marker, in contrast to the CD163 characteristic for the KC [220]. They are highly inflammatory, secrete a variety of cytokines (TNF-α, IL-1β, IL-6, TGFβ) [211] and reach the liver through the CCL2/CCR2 pathway [211]. During obesity, the excess of lipids in the AT promotes lipotoxicity, leading to liver damage and macrophage infiltration [201, 202, 221]. Again, the dietary composition of lipids may play an important role in this context, with omega-3 FFAs displaying interesting anti-inflammatory properties [222–224]. Along with the KC, these macrophages have been indicated as mediators of hepatic inflammation during obesity. The role of liver macrophages in the etiology of obesity/T2DM was established upon depletion of KC and macrophages in the liver resulted in prevention of steatosis, insulin resistance and inflammation [225].
Apart from the macrophages, neutrophils are also recruited to the liver during obesity [73] and contribute to the inflammatory process. As in the AT, the release of neutrophil elastase in the hepatocytes leads to impairment of insulin sensitivity through IRS-2 degradation [73]. Neutrophils together with other cells such as infiltrated monocytes, endothelial cells, fibroblasts, mesenchymal cells, dendritic cells, and hepatocytes produce interferon gamma-induced protein 10 (IP-10), a proinflammatory cytokine associated with the presence of excess fat in the liver [226]. Liver lymphocyte imbalances during NAFLD and NASH were carefully described previously [227]. However, their role in the progression or development of obesity and T2DM has not yet been clearly defined. Decrease in CD4+ T cells [228], and increase in CD8+ T cell and NKT were linked to NAFLD and liver damage [229]. In addition, a gut-liver-intrahepatic CD8+ T cell axis was suggested [230]. This axis was demonstrated to have type 1 interferons as main drivers which provided a mechanism that could be the mediator between alterations in the gut microbiota and subsequent impairment in the insulin action and glucose metabolism during NAFLD and obesity [230].
Concerning the involvement of TLRs in the liver immuno-metabolic response, it is known that the lack of TLR4 protects mice from diet-induced insulin resistance and inflammation [186], and TLR4-deficient hepatocytes were suggested to be responsible for this effect. Mice with TLR4-deficient hepatocytes (Tlr4LKO C57BL/6) showed improvement in both insulin sensitivity and glucose tolerance in addition to steatosis amelioration after exposure to HFD [135].
Despite its central role, the lipotoxicity itself is not the only mediator of the hepatic inflammation. The rate of hepatocyte cell death also plays a role in the resulting proinflammatory environment during obesity and NAFLD. As a consequence of lipotoxicity, a significant loss of hepatocytes due to cell death was observed in the liver [231]. In this context, DAMPS are released and activate inflammasomes [232] which are critical in the progression of NAFLD to NASH. Liver inflammation that leads to disturbed hepatic insulin signaling was also described in wild type rats that after 9 weeks fed a high fructose diet displayed increased inhibitory phosphorylation of IRS-1, increased TNF-α gene expression, enhanced activation of NF-κB [233]. Finally, a recent study demonstrated that liver macrophages produce a non-inflammatory factor named insulin-like growth factor-binding protein 7 (IGFBP7) that regulates liver metabolism [234]. It is suggested that activation of KC to a M1 phenotype does not seem to be required for the development of metabolic disease, indicating that the liver inflammation is, rather, resulted from the monocyte-derived macrophages. These new data places the liver macrophages as strategic direct therapeutic targets in metabolic disease [234].
Pancreas
Composed of various cell types, the pancreas is divided in two distinct functional parts: 1) the exocrine pancreas, which secretes digestive enzymes that break down carbohydrates, lipids and proteins [235], and 2) the endocrine pancreas, that is a source of hormones such as insulin and glucagon which regulate glucose homeostasis [236]. Endocrine cells form pancreatic islets that, in humans, comprise of around 30% of α-cells (glucagon production), 60% β-cells (insulin production), and 10% of δ-cells (somatostatin production) and PP-cells (pancreatic polypeptide production [237, 238]. Immuno-metabolic interactions affect and modulate several internal processes within the islets, particularly concerning β-cells. These cells can sense plasma glucose concentration changes. Glucose is the major and sufficient stimulous for insulin secretion [239]. Failure of the glucose-stimulated insulin secretion is a hallmark of the development of T2DM and an important event in obesity-related conditions [240].
During obesity and metabolic malfunction where insulin resistance is present, β-cells undergo adaptations that stimulate their secretory activity in order to maintain metabolic homeostasis [241]. When those adaptations are insufficient, the excessive overload and demand for insulin lead to saturation and β-cell dysfunction [242]. Different mechanisms have been suggested to explain the β-cell failure in context of metabolic syndrome and T2DM, including, induction of oxidative stress, ER stress and mitochondrial dysfunction as well as imbalance in arachidonic acid metabolism [243–249]. These stress responses were related to mild islet inflammation, that was detected in pancreatic section of T2DM patients [250]. From the immunometabolic point of view, the three following mechanisms may participate in the inflammatory response within islets: 1) macrophage infiltration and/or activation of the tissue resident macrophages in pancreas and the generation of proinflammatory mediators [251], 2) the JNK activation and massive ER stress [176, 252], and 3) intra-islet islet amyloid polypeptide (IAPP) deposits formation and activation of inflammasomes [253–257]. All of them were excellently reviewed in [258]. Exposure to 30 mM glucose of human EndoC-βH1 β-cells did not stimulate IL-1β gene or protein expression [259, 260]. Therefore, it remains controversial whether or not pancreatic β-cells can produce IL-1β, though one cannot exclude the possibility that the T2DM environment with a uniquely composed mixture of various proinflammatory and nutrient factors may induce cytokine production or maturation within β-cells in vivo. Though the anti-IL-1β targeted therapeutic approaches for T2DM resulted in incons is tent outcomes in terms of pancreatic β-cell function protection [261–263], they were shown to reduce serum CRP levels and to promote cardioprotection [257]. Other anti-inflammatory interventions resulted in rather modest protection as discussed in detail in [250].
Similar to other tissues, macrophages infiltration was shown to be increased in islets during T2DM and obesity [264, 265]. Evidence shows that the number of macrophages positively correlates with the severity of pancreatic dysfunction [265, 266] and that infiltrating macrophages exhibit a proinflammatory phenotype [267], suggesting a role for these cells in the progression of β-cell failure. At the same time, another study placed monocyte-derived macrophages as responsible for these events [268], while yet other reports demonstrated that instead resident macrophages play a role [269]. A recent study unraveled the phenotypes and functional specifications of these immune cells in the islets [270]. The authors state that during obesity the islet inflammation is dominated by macrophages, and emphasize the role of the islets-resident macrophages in the immunopathology of the β-cell failure. Immunostaining and RNA-sequencing of pancreatic islets from obese and lean mice showed intra- and peri-islet resident macrophages, being the islets from obese animals rich in CD11c+ macrophages (intra-islet macrophages). Functionally, these intra-islets macrophages were shown to diminish β-cell insulin production and to engulf insulin secretory granules contributing to insulin secretion impairment [270]. Although well-known by a negative impact on glucose-stimulated insulin secretion, a recent study showed that intra- and peri-islet macrophages populations from obese mice stimulated β-cell proliferation in a mechanism dependent on the platelet-derived growth receptor (PDGFR) [270]. Potential differences in the subtypes of islets-macrophages that might play this dual role have not yet been described, as well as whether this feature can be found also in human islets.
Interestingly, the classical inflammatory response pathway of arachidonic acid metabolism is present in β-cells and undergoes significant changes under diabetogenic conditions [246–249]. Pancreatic β-cells are characterized by an imbalance in the expression profile of enzymes involved in the arachidonic acid cascade with the weak expression of prostacyclin synthase expression, responsible for the generation of the anti-inflammatory prostacyclin (prostaglandin I2) [249–271]. The proinflammatory prostaglandin E2 was shown to reduce glucose-induced insulin secretion [248]. In contrast, the anti-inflammatory prostacyclin is a strong potentiator of insulin secretion [249]. Prostaglandin synthesis inhibitors [272] as well as prostacyclin analog beraprost sodium [273] were shown to ameliorate characteristics of metabolic syndrome in obese Zucker fatty rats and to improve insulin secretion in diabetic patients, respectively.
Recent studies evaluated the protective potential of omega-3 FFAs for lowering of inflammation via formation of SPMs in T2DM and obesity animal models and diabetic patients with various outcomes [25, 274]. While the role of SPMs in islet inflammation in T2DM remains currently unknown, SPMs have been shown to promote M2 polarization of macrophages, reduce AT and muscle inflammation, increase insulin sensitivity and lower fasting blood glucose in diet-induced obese mice, ob/ob mice or obese-diabetic mice [26, 275–277].
Further investigations are needed to better elucidate the immuno-metabolic interactions within pancreatic islets and to explore the therapeutic potential of anti-inflammatory metabolites of arachidonic acid cascade.
In conclusion, immunometabolism is an emerging field that investigates the relationship between metabolic and inflammatory processes. These investigations are of special interest to clarify how metabolic disorders develop and progress. Inter- and intra-organ interactions were presented using the most updated reports in the field and summarized in Figure 1. The review indicates that many studies are still needed to uncover the molecular mechanisms behind this cross-talk that influences central organs responsible for the whole-body homeostasis.
T2DM and obesity are disorders in which inflammation plays an important role in the pathogenesis. How does inflammation become so harmful to the point of causing or worsening metabolic disorders? The discussed data brings the innate immune cells, especially macrophages, as big players in secreting proinflammatory factors that directly impair metabolic tissue functions. Besides some particularities, the adaptive immune response (consequently activated) and proinflammatory pathways such as JNK and NF-κB are also important contributors to the inflammatory environment in multiple organs. In the AT, B-regs and cytokines, such as IL-37, represent potential therapeutic targets. In the gut, the modulation of the intestinal barrier permeability and clarifications of the impact of dysbiosis will bring important discoveries. In the liver, much is still mysterious about the impact of its resident macrophages. In the muscle the intriguing dysregulation of myokine production and formation of intramuscular fat depots require further investigations. Finally, mild, but persistent, and largely still under investigated inflammation of pancreatic islets may open new therapeutic possibilities to preserve proper β-cell function.
Besides many exciting and promising discoveries that unravel mechanisms particularly important to obesity and diabetes, it is important to emphasize a challenge: the translation of the knowledge to the human situation and the arising limitations. Inter-individual variability in humans influences the type of immune response and its magnitude [278]. It translates into a major challenge in the development of therapies that do not harm the immune system as a whole. Cross studies, genetic variations, and environmental considerations will be prerequisite to make the translation a reality. Finally, the role of inflammation in the maintenance of homeostasis and protection against infections and injuries should not be neglected. Despite the elucidation of new players and their interactions in the immuno-metabolic crosstalk, translating them to therapeutic targets may compromise the body’s ability to defend itself. Future investigations should uncover not only new mechanisms involved but also provide answers on how to apply them in order to treat and cure metabolic diseases.