Introduction
IgG4-related disease (IgG4-RD) is a set of signs and symptoms resulting from a chronic, usually multiple organ inflammatory condition which affects various tissues and organs. It consists of lymphoplasmacytic infiltrations with attendant fibrosis and deep vein thrombosis [1]. Frequently observed tissue lesions are accompanied by elevated IgG4 levels in serum. The etiopathogenesis of the lesions is of multifactor character and includes allergy mechanisms as well as infectious and autoimmune factors. Therefore, medical signs and diagnostics pose a serious challenge to clinicians. The epidemiology of IgG4-RD has not been fully clarified yet. The majority of cases are reported in Asia, though apparently IgG4-RD may occur all over the world [2]. According to Japanese researchers, the average number of newly diagnosed patients is 0.28–1.08/100 000 in the whole population [3]. In the medical literature different names have been employed to describe IgG4-RD. Only after a consensus had been reached by two independent Japanese research committees was the currently used name finally acknowledged [4]. The systemic character of IgG4-RD was first observed in 2003, mainly with reference to autoimmune pancreatitis [5]. Since then, almost forty different locations of the disease have been reported, including disorders of the endocrine system [2]. The IgG4-related endocrinopathies are quite rare, and thyroid involvement is believed to occur in 4% to 6% of cases. However, it is likely that the diagnosis is under-reported due to lack of awareness of this clinical entity. Despite an increasing interest in the subject, there are not enough reliable studies evaluating the link between IgG4-RD and endocrine disorders.
Epidemiology
Men are usually reported to suffer from IgG4-RD more frequently than women although in the case of involvement of head and neck organs these proportions become equal [6, 7]. Mean age at the onset of the disease is between 50 and 60 years [2, 8]; however, the disease may occur at any age, even in children [9, 10].
Most frequently, the lesions are located in organs of the digestive system such as the pancreas, bile ducts and salivary glands, as well as lacrimal glands, soft tissues of the orbital cavities and lymph nodes. Less frequently the disease may also be located in the mediastinum, respiratory system, retroperitoneal space, central nervous system, genitourinary system, mammary gland, prostate gland, cutis and blood vessels [2, 3]. Among the most frequently infected endocrine glands are the thyroid and pituitary. Table I presents the most common organ locations in three large case series.
Table I
Comparison of organ involvement in three large case series
| Organ involvement | Wallace et al. [6] Percentage of 125 cases | Inoue et al. [82] Percentage of 235 cases | Ebbo et al. [83] Percentage of 25 cases |
|---|---|---|---|
| Salivary gland | 34 | 44 | |
| Submandibular glands | 28.0 | ||
| Parotid glands | 16.8 | ||
| Lacrimal gland | 23 | 12 | |
| Pancreas | 19.2 | 60 | 52 |
| Bile duct/biliary | 9.6 | 13 | 32 |
| Orbital cavity | 22.4 | 4 | 4 |
| Lungs | 17.6 | 13 | 12 |
| Kidneys | 12.0 | 23 | 44 |
| Aorta | 11.2 | 20 | 24 |
| Lymph nodes | 27.2 | 76 | |
| Intraperitoneal fibrosis | 18.4 | 4 | 32 |
| Pituitary gland | 8 | ||
| Thyroid | 5.6 |
Pathophysiology of IgG4
The pathogenesis of IgG4-related disease is not fully elucidated. A combination of mechanisms of the innate and adaptive immune system, as well as genetic factors, contributes to the development of the disease. Most studies so far have examined only Asian patients with autoimmune pancreatitis (AIP), a pancreatic manifestation of IgG4-related disease.
The role of IgG4
Immunoglobulin G4 is one of the factors contributing to the development of IgG4-RD. However, its role in this complex process has not been fully explained yet. It has been established that in physiological conditions IgG4 constitutes the lowest IgG percentage in serum (3–6%). It is characterized by unique properties, such as greater sensitivity to chemical factors as well as incapability of creating immune complexes [11, 12]. It has been demonstrated that IgG4 production depends on T2 lymphocytes, which participate in allergization processes. An IgG4-related physiological response may also be triggered by a repetitive exposure to an antigen, e.g. in food allergy [13]. Although IgG4 does not have the ability to activate the classical complement pathway, it contributes to an inflammatory response through histamine release from basophils [14], which may partially account for its elevated levels in patients with atopic diseases and clinical improvement following the elimination of food allergens from the diet [15]. In the medical literature there are reports regarding the “inhibiting” character of the IgG4 antibody. Van der Giessen et al. observed a high concentration of this immunoglobulin following successful immunotherapy of IgE-related pollen allergies [16].
Elevated IgG4 levels may be observed not only in patients with IgG4-RD (in approximately 60–70% of patients), but also in a chronic inflammatory condition, infections, cancer, autoimmune diseases, vasculitis and drug hypersensitivity.
Pathomechanism
Several groups have tried to identify autoantigens based on the hypothesis that IgG4-related disease is an autoimmune disorder. The factor which triggers the disease process in IgG4-RD is still being searched for. Until now, a few antigens have been identified, including lactoferrin, carbonic anhydrase (CA-II), and pancreatic secretory trypsin inhibitor (PSTI) [17, 18]. Recent publications suggest a possible role of prohibitin as an antigen which induces the generation of antibodies that contribute to the tissue growth and development of the IgG4-RD [19]. Others, on the other hand, indicate a possible role of molecular mimicry with the participation of Helicobacter pylori [20].
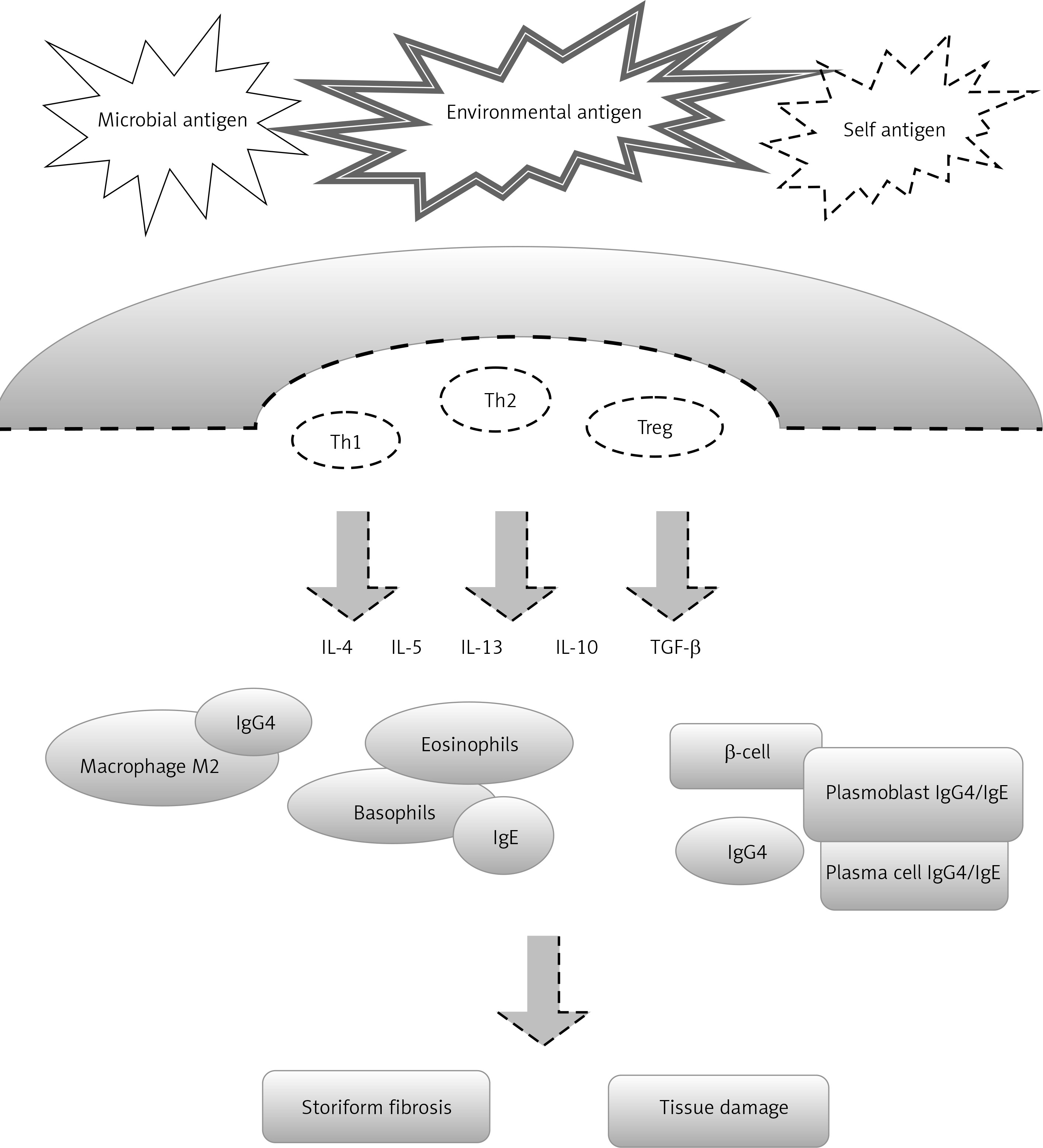
Increased tissue levels of Th2-cytokines including IL-4, IL-5, IL-10 and IL-13 were found in IgG4-RD. These cytokines also trigger antibody class switch to IgE and eosinophilic migration and infiltration. A role for elevated levels of regulatory T cells (Treg) has also been suggested. Two regulatory cytokines, IL-10 and transforming growth factor-β (TGF-β), were also significantly overexpressed. Il-10 directs B cells to produce IgG4, while TGF-β, a key pro-fibrotic cytokine, induces the deposition of extracellular matrix proteins and type 1 collagen, leading to fibroplasia. Further, 20% of IgG4-RD patients have allergic diseases (e.g. bronchial asthma or drug allergy), and tissue eosinophilia is typical [21]. However, Della Torre et al. stated that the majority of patients with IgG4-RD are nonatopic and that the prevalence of atopy in this disease is no higher than that expected in the general population [22]. A study showed that there are increased numbers of M2 macrophages in submandibular glands affected by IgG4-RD and that fibrosis is associated with an increased number of M2 macrophages [23].
The B cell compartment of patients with IgG4-RD has been studied extensively, because IgG4 elevation in the serum and the abundance of IgG4-plasma cells in the biopsies initially suggested an underlying lymphoproliferative condition. In B-cell development, plasmablast differentiation occurs between activated B cells and plasma cells. Wallace et al. reported that circulating plasmablasts are elevated in active IgG4-RD, even in patients with normal serum IgG4 concentrations, and that their levels reflect disease activity [24].
In the IgG4-RD pathomechanism, there have also been detected certain abnormalities in cell-mediated immunity. Monocyte and basophil disorders with the overproduction of cytokines such as BAFF and APRIL have been reported [25].
There is some evidence from Japanese studies that certain HLA alleles – particularly DRB1*0405 and DQB*0401 – increase the susceptibility to type 1 autoimmune pancreatis (AIP). Furthermore, a few single-nucleotide polymorphisms (SNP) in genes related to the immune system such as CTLA-4, Fc-receptor-like 3 have been linked to IgG4-RD [21].
Figure 1 presents a simplified diagram of the IgG4-RD pathomechanism.
Clinical manifestation
Clinical manifestation of the disease is highly diverse, and its course in the initial stage is often asymptomatic. Therefore, the appearance of the symptoms seems to depend on the stage of the disease process, the number of infected organs and the duration of the disease. During the development of the disease, diffuse or focal organ enlargement may be observed, together with pseudotumours, erosion and sclerosis of adjoining bones [7]. In approximately 60% of patients at the moment of the diagnosis the disease process is already of multiorgan character. One third of the patients have a positive medical history for atopic diseases, which refers mostly to the respiratory system, cutis and soft tissues of orbital cavities [22]. General symptoms, such as fever, weight loss, and asthenia, are not frequently observed. Lymphadenopathy is reported in nearly 42% of patients with a systemic form of the disease. The appearance of specific symptoms depends on the location of the disease process and involves abdominal pain, keratoconjunctivitis sicca, various respiratory disorders, pruritus and diarrhea [26].
Diagnostics and its criteria
The diagnostic process is based on the patient’s medical history, clinical examination and additional tests, including a histopathological examination of the infected organ’s tissues [27]. Umehara et al. have established clinical criteria which determine the likelihood of IgG4-RD diagnosis (Table II) [4].
Table II
Criteria for the diagnosis of IgG4-RD according to Umehara et al. [4]
The microscopic image of the infected tissue is typically characterized by dense infiltrations formed by lymphoplasmacytic cells (B and T lymphocytes), storiform fibrosis and thromboangiitis obliterans. Eosinophilic infiltrations are sometimes reported, especially in lesions located in orbital cavities or the upper respiratory tract [28].
Upon suspicion of IgG4-RD, it is necessary to perform immunohistopathological tests in order to identify IgG4+ cells and evaluate the IgG4+/IgG ratio, which in the case of IgG4-RD diagnosis should be higher than 0.4 [4, 28].
So far it has been established that there may be a correlation between IgG4 concentration in serum and the activity of IgG4-RD. High levels of this immunoglobulin are usually associated with a more aggressive course of the disease, concurrence of IgG4-related lesions in other organs and probably a reduced response to the treatment [27]. What hinders the diagnostic process to a certain extent is the fact that IgG4 concentration may be within the reference range even in half of the patients with an active form of the disease confirmed in histopathological tests [6]. Therefore, isolated elevated IgG4 levels do not determine the diagnosis.
Abnormalities in laboratory tests are not very specific and include moderately increased inflammation markers (CRP in 25% of cases), mild eosinophilia (in 34% of cases), and sporadically erythrocytopenia and thrombocytosis [18]. Furthermore, there are visibly elevated IgE levels [15], polyclonal hypergammaglobulinemia [29], decreased levels of complement components C3 and/or C4 (21% of cases, especially in IgG4-related renal disease) and rheumatoid factor and a low anti-nuclear antibody index [30].
Medical imaging facilitates the diagnosis, yet it is not specific enough to differentiate between IgG4-RD and inflammation or a hyperplastic process. It is believed that the main role is played here by computed tomography, magnetic resonance imaging [26] and, above all, FDG PET-CT [31].
Treatment methods
Beginning the treatment immediately after the diagnosis of IgG4-RD is not always necessary. In the case of oligosymptomatic forms of the disease, with a mild course, the treatment may be postponed on condition that the patient is carefully monitored (“watchful waiting”).
Until now there have been no unified directives established regarding the treatment of IgG4-RD. Glucocorticoid administration is the first-choice therapy in patients with an active, previously untreated form of IgG4-RD. According to the directives from international guidelines, the common initial dose of prednisone (0.6 mg/kg body weight/day, 30–40 mg/day) should be administered for 2–4 weeks, and then gradually reduced. One scenario is to taper the daily dosage by 10 mg every 2 weeks until a daily dosage of 20 mg is reached. After a short time (e.g., 2 weeks) of treatment at 20 mg/day, the tapering should resume by decreasing the daily dosage by 5 mg every 2 weeks. The patient should terminate the treatment after a period of 3–6 months. However, some Japanese findings suggest that administration of small prednisone doses (2.5–5 mg/day) may be continued up to three years. In remittent and treatment-resistant forms of the disease studies have shown the efficacy of certain immunosuppressive drugs (azathioprine, 6-mercaptopurine, methotrexate, tacrolimus, cyclophosphamide) and biological compounds (rituximab) [27].
Treatment of IgG4-related endocrinopathies is believed to be the same as in patients with IgG4-related disease, but their use in management needs to be evaluated.
Risk of neoplastic disease
According to some findings, the risk of cancer increases threefold in patients with IgG4-RD in comparison to the general population [32]. The medical literature describes cases of non-Hodgkin lymphoma as well as lung carcinoma, pancreatic cancer, stomach cancer, bile duct cancer, kidney cancer, breast cancer, tongue cancer, melanoma and acute myeloid leukemia [33]. There have been reported single cases of concurrence of Riedel’s thyroiditis and thyroid cancer [34].
Thyroid disorders associated with IgG4-related disease
The concept of IgG4-related thyroid disease was first proposed in 2011. It was then observed that 19% (22 out of 114) of patients with IgG4-related diseases present clinical markers of hypothyroidism. It was observed that glucocorticoid administration in 10 patients with clinically symptomatic hypothyroidism resulted in a statistically significant improvement in their hormonal parameters [35].
So far, on the basis of existing medical publications, thyroid disorders associated with IgG4-RD may be divided into Riedel’s thyroiditis (RT), IgG4-related Hashimoto thyroiditis, (IgG4-RHD), fibrous variant of Hashimoto thyroiditis (FVHT), and Graves’ disease related to elevated levels of IgG4 [36].
Riedel’s thyroiditis is a very rare disease, with a frequency of 1.06/100 000 in the whole population [35]. Contrary to IgG4-RD, Riedel’s thyroiditis occurs more frequently in women than in men (3 : 1). The peak age of incidence is between 30 and 60 years. The etiopathology of the disease has not been fully clarified. Most common in clinical manifestation is massive fibrosis of the thyroid gland and the surrounding tissues [37]. In clinical examination the thyroid is enlarged, painless and extremely hard, fixed to adjacent structures. Both in clinical examination and during surgery, it is difficult to distinguish between Riedel’s thyroiditis and thyroid anaplastic cancer or lymphoma. Moreover, patients may have symptoms of tracheal and esophageal pressure (dysphonia, dyspnea, stridor, dysphagia), monolateral or bilateral paralysis of the recurrent laryngeal nerve, hypoparathyroidism, Horner’s syndrome, or superior vena cava syndrome. A few dozen percent of patients present high levels of anti-thyroid antibodies, yet their role in pathogenesis of the disease remains unknown [38]. Schwaegerle et al. observed elevated levels of anti-thyroid antibodies in 67% of patients with RT. Upon diagnosis most patients are euthyroid, and approximately one third of them develop hypoactivity of the gland [39]. The microscopic image in RT presents similar results as the typical image of IgG4-RD and serves as the basis for the diagnostic and therapeutic process. In 2010 Dahlgren et al. reported cases of 3 patients with RT, using immunohistochemical tests to demonstrate the presence of IgG4+ cell infiltrations in the thyroid gland [40]. On the basis of later research RT has been included in the IgG4-RD spectrum [1].
The choice of treatment in patients with Riedel’s thyroiditis depends primarily on the stage of the disease. In case of hypothyroidism, among the drugs, along with L-thyroxine supplementation, an important role is played by glucocorticoids. Glucocorticoid starting doses range widely from 15 to 100 mg/day, and are generally reduced slowly over weeks to months. Remission rates after glucocorticoid usage are unclear, but relapse can occur during dose reduction. Nonsmokers respond better than smokers [41].
Tamoxifen is the second-line agent which has been used successfully in patients who relapse on glucocorticoids. Doses of 10–20 mg of tamoxifen either in combination with continued glucocorticoid therapy or as a monotherapy have been reported to be successful, especially in symptom control and reduction of pathological tissue mass [42].
In treatment-resistant patients mycophenolate mofetil and rituximab are of great importance for RT treatment [43, 44].
Hashimoto disease (HD) is the most common autoimmune disease and at the same time the most common cause of hypothyroidism in iodine-sufficient areas. The incidence of this disease increases with age (over 17% above the age of 60) and is much higher in women than in men (7 : 1). Most common in a typical clinical manifestation are: painless, diffuse goiter of increased density, elevated levels of anti-thyroid antibodies and hormonal markers of hypothyroidism, especially at the more advanced stage of the disease. There are a few clinical variants of HD [45], one of them being IgG4-related Hashimoto thyroiditis (IgG4-RHD). According to Chinese studies, the incidence of IgG4-RHD among patients with HD is 22.64% (12 out of 53 cases) [46]. Similarly to IgG4-RD, the medical manifestation of this disease is characterized by fibrosis with lymphoplasmacytic infiltrations and follicular cell degeneration, which rapidly lead to hypothyroidism [47, 48]. Additional examination shows high levels of anti-thyroid antibodies and diffuse hypoechogenic lesions in ultrasonographic imaging [49].
Recognized since 1974, fibrous variant of Hashimoto’s thyroiditis (FVHT) appears to be included in IgG4-RHD; however, there are no unified findings distinguishing these two disease entities [50]. Contrary to RT, there is so far no clinical proof of the efficacy of glucocorticoids in HD treatment [51]. Table III presents clinical markers distinguishing RT from FVHT [52].
Table III
Clinical markers for distinguishing between Riedel’s thyroiditis and fibrous variant of Hashimoto disease
During the research into the role of IgG4 in the diagnostics of autoimmune thyroid diseases, it has been observed that it may be used as a potential marker of disease activity. Elevated IgG4 levels in serum are reported in up to one third of patients with HD. This group develops a more aggressive course of the disease, requiring higher doses of L-thyroxine [53], although there are publications which contradict this observation [54].
Similar correlations have been searched for in patients with Graves’ disease, yet it has been observed that only a small group of patients (6.4%) are “IgG4-positive”. However, the group’s unique characteristics, such as proneness to hypothyroidism following the antithyroid therapy and greater tendency to develop Graves’ ophthalmopathy, necessitate conducting further research in this field [55].
On the basis of currently available findings, it seems that IgG4-related thyroid disease must be considered in patients with a clinically aggressive form of the disease, and an immunohistochemical test with IgG4/IgG assessment should be performed in all of those patients.
IgG4-related pituitary disease
Primary inflammatory conditions of the pituitary gland are relatively rare, accounting for only 0.5% of all causes of hypoactivity of this gland [56]. Until quite recently, there were four basic forms of histopathological inflammation: lymphocytic (most frequent), granulomatous, xanthomatous and necrotizing. One of the newly discovered forms is IgG4-related hypophysitis (IgG4-RH), which accounts for merely 1.5% of systemic cases of IgG4-RD [2]. The first publications regarding this disease entity appeared in 2004 and described a case of a 66-year-old woman with numerous pseudotumorous lesions in the salivary glands, pancreas and peritoneal cavity, with concurrent secondary hypothyroidism and sella turcica enlargement in tomographic imaging of the pituitary gland [57]. Further studies systematized the clinical and radiological manifestation of this condition. Similarly to IgG4-RD, the incidence is higher in men than in women, and the mean age of onset is 64 years. General weakness, headaches and vision disorders are predominant in clinical examination. Other symptoms include polyuria, weight loss, high body temperature and decreased libido. Also observed are various degrees of hypoactivity of the anterior lobe of the pituitary gland and central diabetes insipidus. Complete pituitary hypoactivity occurs in 52.4% of the described cases, and as many as 75% of patients develop antidiuretic hormone deficiency [58]. Magnetic resonance imaging of the pituitary is not very specific, mostly showing edema or pseudotumorous enlargement of the gland. A “bright spot” in the posterior lobe may be observed in patients with central diabetes insipidus. Similar lesions also occur in other inflammatory conditions of the pituitary gland (e.g. lymphocytic inflammation), as well as in sarcoidosis, tuberculosis and Wegener’s granulomatosis [59].
In 2011 Leporati et al. proposed three diagnostic criteria of IgG4-related pituitary disease, the fulfillment of which is sufficient for establishing the diagnosis. The first criterion requires a microscopic examination of the pituitary gland, with confirmed presence of mononuclear cell infiltration with over 10 IgG4 cells in the field of vision. The second criterion is atypical manifestation of pituitary gland inflammation visible in MRI and confirmed infiltrations located elsewhere. The last criterion is the concurrence of elevated IgG4 levels in serum, manifestation of pituitary inflammation in MRI and rapid symptom remission following glucocorticoid administration [60].
The diagnostics of IgG4-related pituitary disease undoubtedly poses a serious challenge for clinicians. The microscopic image and the result of immunohistochemical examination may not differ significantly from other variants of pituitary inflammatory conditions. Lymphocytic infiltrations of IgG4 cells have also been described in connection with other diseases [61]. A diagnosis for IgG4-RH should be considered in patients who develop meningitis or have a relapse induced by reduction of the glucocorticoid dose [62].
The typical therapy for IgG4-related hypophysitis is undefined. It is noteworthy that there are clinical cases in which high clinical efficacy of small glucocorticoid doses is reported from the beginning of treatment [63, 64].
IgG4-related disease versus focal adrenal lesions
Focal adrenal lesions regarded as IgG4-related are extremely rare. The only example of this condition confirmed so far is calcifying fibrous tumor that belongs to a group of benign mesenchymal cancers, which usually develops in the stomach, lungs, mediastinum, peritoneum, and in approximately 3% of cases also the adrenal glands [65]. A classic example, described in 2016, is a case of a 32-year-old patient with a lesion in the left adrenal gland accidentally detected during an ultrasound examination. The medical picture was asymptomatic and hormonal test results were within the reference range [66].
IgG4-related ophthalmic disease
Graves’ orbitopathy is a frequent clinical problem in endocrine practice, affecting on average 25% of patients with Graves’ disease [38]. In differential diagnosis of this condition a range of diseases has to be taken into consideration, with IgG4-related ophthalmic disease recently classified as one of them [67].
The first findings regarding IgG4-RD with infection of the orbital soft tissues were described in 2011 in Italy, when Fonte et al. presented a case of a patient with autoimmune pancreatitis and Graves’ orbitopathy, with attendant high IgG4 levels in serum and euthyroid. The clinical course of the disease was similar to an active, acute or moderate form of Graves’ ophthalmopathy. A good response to intravenous glucocorticoid administration and radiotherapy of the orbital cavities was observed [68]. On the basis of further studies the term “IgG4-related ophthalmopathy” was proposed [1].
In the clinical picture the disease is usually bilateral and is characterized by a mild course. Pain sensation in the eyes or visual field disorders do not occur frequently. Most common in clinical examination are chronic eyelid edema and exophthalmos, with uninhibited eye movement. There is a possibility of infection of the lacrimal gland (70%), orbital bone structure, ophthalmic nerve or a branch of the trigeminal nerve (V1 and V2). Extraocular muscles are extended in one third of the patients. The most common location is the lateral rectus muscle [69–71]. Sometimes there can be observed symptoms resulting from infections of other organs such as submandibular glands (29%), lymph nodes (14%), pancreas (5%) and bile ducts (5%) [71–74]. Similarly to other forms of IgG4-related diseases, a histopathological examination is the basis for the diagnostic process. In the microscopic image lymphoplasmacytic and eosinophilic infiltrations prevail, whereas vein thrombosis is very rare [74]. The core of the therapy is glucocorticoids, which in approximately 70% of patients should be administered long-term in monotherapy or in conjunction with other immunosuppressive drugs. Biopharmaceuticals such as rituximab or bortezomib are suitable for patients with a treatment-resistant form of the disease [27, 71]. The recommended dose of rituximab is two pulses of 1.0 g with a 2-week interval. Patients may require more than one cycle of rituximab, or may experience relapse after 6 months when peripheral B-cells are re-populated [75].
A diagnosis of IgG4-RD should be considered in the differential diagnosis of orbitopathy of unclear etiology, especially treatment-resistant and in cases of concurrent symptoms in other systems.
Diabetes in the course of autoimmune pancreatitis
Autoimmune pancreatitis is a syndrome consisting of general symptoms, obstructive jaundice and often concurrent carbohydrate imbalance. There are two types of AIP. Type 1 AIP (IgG4-related pancreatitis) is more prevalent in Japan and Korea, whereas type 2 AIP, with granulocytic epithelial lesions, is more commonly observed in Europe and the United States [76].
AIP-related diabetes may occur at every stage of the disease. However, in more than 50% of patients it is reported at the moment of the AIP diagnosis. It is remarkable that the symptoms of AIP-related hyperglycemia may completely disappear following glucocorticoid administration [77]. Around 60% of cases of diabetes mellitus associated with AIP are responsive to steroids in the short and long term [78]. Yamada el al. described a case of a 79-year-old woman whose diabetes was the only AIP manifestation, and glycemic balance after the first month of steroid therapy was maintained without hypoglycemic drug administration [79]. In the majority of cases, “IgG4-related diabetes” is treated with insulin therapy and its course depends on how advanced the pancreatic atrophy is [80–83].
Conclusions
IgG4-RD is a new, multidisciplinary condition, which poses a challenge to clinicians. The scope of this phenomenon and its role in contemporary diagnostics appear to be a subject which truly deserves further research as well as medical experiments. Thyroid and pituitary gland diseases constitute the highest percentage of IgG4-related endocrinopathies.