The mammalian gastrointestinal tract is continuously vulnerable to numerous bacteria and food additives as well as to environmental toxins that damage the epithelia of the gut [1]. Disturbance in intestinal homeostasis triggers a pathogenic immune response commonly known as inflammatory bowel disease (IBD) [2]. IBD is a complex idiopathic disorder including clinical Crohn’s disease (CD) and ulcerative colitis (UC), which are characterized by chronic inflammation in the mucosal layer of the gut, which sometimes affects the whole intestine with numerous etiological manifestations [3, 4]. IBD is clinically manifested with diarrhea and rectal bleeding accompanied by mucous pus [5] and is commonly associated with childhood, which includes symptomatic manifestations such as abdominal cramps, and excessive weight loss [6]. A large number of people annually are diagnosed with IBD, a major health issue in western societies that affects around 1.4 million patients with an incidence rate of 29.6 in 100,000 which includes clinical complexities with poorly effective treatment regimens [7]. Moreover, multiple comorbidities that exaggerate IBD pathogenesis include genetic predisposition, immunological disorders, a high-fat diet, and regular use of antibiotics [8].
The IBS pathophysiology is still unclear but involves the disruption of the mucinous layer, which serves as the primary defense line of the gut immune system and provides a physical barrier between the immune cells of the host and synthesis of antimicrobial peptides. Additionally, levels of colonic mucin-2 are decreased and altered during IBS [9]. This damage further facilitates increased permeability of epithelium of gut mucosa and alters the tight junctions, which ultimately lead to the uptake of luminal antigens [10].
The animal model for IBS exhibits many similar characteristics to human studies, which include diarrhea-associated inflammation, weight loss, immune cell infiltration, and activation with altered macrophage functions [11]. The most suitable and commonly used animal model to study experimental colitis is induced by administering dextran sodium sulfate (DSS), a chemical colitogen with anticoagulant properties, to understand IBD pathogenesis [12]. DSS is a toxic reagent that triggers damage in the colonic mucosa and provokes an inflammatory response [6]. DSS-induced intestinal inflammation in IBD is unclear but its multi-mechanistic pathological cascade is linked to disruption of the monolayer lining in the large intestine with distribution to the rectum and entire colon [12]. Moreover, it disrupts the epithelial layer of the colon, which recruits inflammatory cells and accelerates the excessive release of pro-inflammatory cytokines such as tumor necrosis factor-α (P-JNK-α), and interleukins 1β and 6, which leads to necrosis [13, 14]. The DSS-induced IBS model in the research holds relevance due to its simplicity, rapidity, reproducibility, and constructability in animal studies, and it signifies the relapse of intestinal inflammation through adjustment of DSS concentration and variability in the frequency of dosage administration [12]. Therefore, IBS induced through DSS is considered a requisite tool in the identification of underlying pathological cascades and pathways in IBS and identification of novel therapeutic targets. Several therapeutic agents have been developed for treatment of IBD. However, the results in efficacy are still unsatisfactory including side effects of systemic medications and complications of drugs [15, 16] which potentiate the need for alternate therapy.
Ibrutinib is a novel therapeutic inhibitor of BTK which is approved for the treatment of several disorders such as chronic lymphocytic leukemia (CLL) and mantle cell lymphoma [17, 18]. It is also involved in the treatment of hematological malignancies such as chronic lymphocytic leukemia, specific lymphomas [19], and Wald Enstrom’s macroglobulinemia. Ibrutinib is recognized as the first BTK inhibitor that has been approved by the food and drug regulatory authority [20]. As per our literature survey, there are fewer studies on the role of ibrutinib in gastrointestinal disorders; therefore in this study we investigated the pathophysiology of DSS-induced IBD and possible protective effect of ibrutinib to attenuate IBD induced necrosis.
DSS (Catalog No: ALDRICH_42867) was purchased from Med Chem Express USA, ibrutinib (Catalog No: W224511) was purchased from Sigma Laboratories (Sigma-Aldrich, United States), mesalazine (Catalog No: PHR10999) from Pharmedic Laboratories, Private Limited Lahore, Pakistan. Phosphate buffer saline tablets and proteinase K (Catalog No: SAB4700839) were obtained from (MP Bio, United States). Formaldehyde, hydrogen peroxide (H2O2), reduced glutathione (GSH), glutathione S-transferase (GSTs), catalase, trichloroacetic acid, CDNB, ethanol, xylene, hematoxylin, and eosin stains were purchased from Sigma-Aldrich, United States. Primary antibody P-NF-κB (SC-271908), p-PI3K (SC-293115), p-AKT (SC-7985), p-GSK3β (SC-11757), p-JNK (SC-6254), TNF-α (SC-52B83), COX-2 (SC-514489) and other immunohistochemistry-related consumables, such as AB and C Elite kit (two vials-SC-2018) and 3,3-diaminobenzidine (DAB) (SC-216567), were provided by Santa Cruz Biotechnology, USA. The biotin secondary antibody (ab-6789) and DPX mounting media were purchased from Abcam UK.
Seventy healthy male adult Albino mice having weight 35 ±15 g, and 8–12 weeks of age, obtained from the local breeding facility of Riphah International University Islamabad, were randomly divided into five groups (14 mice/group). The mice were placed in plastic cages and held at room temperature 18–22°C, with a light/dark cycle of 12 h. Throughout the experiment, animals were given free access to rat pellet diet ad-libitum and tap water. For the entire duration of the research, animals were kept under continuous surveillance. To keep the mice free from stress, all appropriate managerial procedures were adopted. Prior approval was obtained for all such experimental protocols from the Research and Ethical Committee (REC) of Riphah Institute of Pharmaceutical Sciences (RIPS), Riphah International University, Pakistan. Mice were randomly divided into five experimental groups, i.e., control group (saline), disease group (DSS-3%), standard group (mesalazine 10 mg/kg), treatment group 1 (ibrutinib 6.25 mg), and treatment group 2 (ibrutinib 12.5 mg), and these doses were selected as in previous literature [21, 22]. A total of 15 animals died during the experimental procedure due to excessive bleeding in feces and dehydration, i.e., 6 from the disease group, 2 from the mesalazine group, and 2 from both ibrutinib groups.
To avoid a potential impact, colitis-induced mice and their littermates were co-housed and acclimatized for over a week. Colitis was induced in mice following the protocol published before [23]. Briefly, mice (male, 8–12 weeks old) fed a normal diet were administered 3% dextran sulfate sodium salt (DSS) (MW 36–50 kDa; MP Biomedical, Irvine, CA, USA) dissolved in drinking water for 10 days. Control littermates were given regular drinking water. During induction, mice were weighed and stools were tested for occult blood daily to assess pathology severity. At the end of induction time points, mice were euthanized by CO2 inhalation. Colon and other tissue were collected and snap-frozen in liquid nitrogen and stored at –80°C for further use. Blood was centrifugated in 1000 g for 30 min at 4°C to isolate serum. The serum was stored at –80°C for further use as well.
The course of inflammation was monitored by weight loss measurements and the stool morphology test. Pathological scoring was used to quantify the severity of colitis following a method adapted from the standard DAI scoring system [24]. Each animal was examined once a day and received a score containing three parameters, body weight decreasing, fecal bleeding, and stool consistency. For the assessment score, the definition was as follows: the decrease in body weight relative to initial weight reached 0–1% scored 0, 1–5% scored 1, 5–10% scored 2, 10–15% scored 3 and over 15% scored 4; the stool normal and no blood scored 0, a dimly visible streak of blood scored 1, moderate blood scored 2, obvious blood scored 3 and gross bleeding from the anus scored 4. As for the stool viscosity, normal, the score of 0; soft, 1; loss, 2; diarrhea, 3 and diarrhea with blood, 4. Then these features were averaged for each mouse and each group to calculate the percentage of diarrhea.
Mice were subjected to fasting condition for 8 h before blood collection. The blood sample was collected on day one via the tail vein to measure the levels of liver enzymes. At the end of the study period i.e., the 10th day, mice were anesthetized and blood samples were collected by direct cardiac puncture using a 3 ml BD syringe. For serum biochemistry, blood was collected in clotted tubes, blood was permitted to clot at 26 ±2°C, serum was extracted by centrifugation at 3000 RPM for 10 min. After obtaining blood samples, all mice were euthanized and the sample was processed.
Blood samples drawn from cardiac punctures, collected in clotted vials, were utilized for estimation of serum biochemical parameters. Biochemical assays of serum were performed using biochemical assay kits (TB, ALT, ALP, AST) on a biochemistry analyzer (Beckman Coulter AU480, USA). All procedures were conducted in compliance with the manufacturer’s protocol.
The collected samples were fixed in 4% formaldehyde and decalcified using nitric oxide. The histological samples after the gross study were embedded in paraffin blocks, cut into 4 µm thin sections using a microtome, and the following analysis was performed.
H& E staining was performed as mentioned in our previous paper. Absolute xylene (100%) was used to de-paraffinize the tissue sections on coated slides, rehydrated with ethyl alcohol from 100% to 70%. Following washing with distilled water, slides were incubated in hematoxylin solution for 10 min (depending upon the nature of the hematoxylin stain). The hematoxylin stain was recycled and slides were put into low-speed tap water, subsequently dipped for one gentle treatment with 1% HCl and 1% ammonia water. The slides were finally incubated with eosin and kept for air drying. The dried slides were processed with the dehydration protocol and mounted with a glass coverslip. Histological assessment was performed under a light microscope for measuring villus heights (from villus tip to villus-crypt junction), crypt depth (invagination depth between adjacent villi), crypt cell apoptosis, and mitotic indexes. Twenty well-oriented villi and crypts for villus height, crypt depth, and mitotic figures per crypt were measured and scored for the average of each sample. All intestinal morphometric assessment was performed blindly by ImageJ software 1.8_172 (NIH, USA).
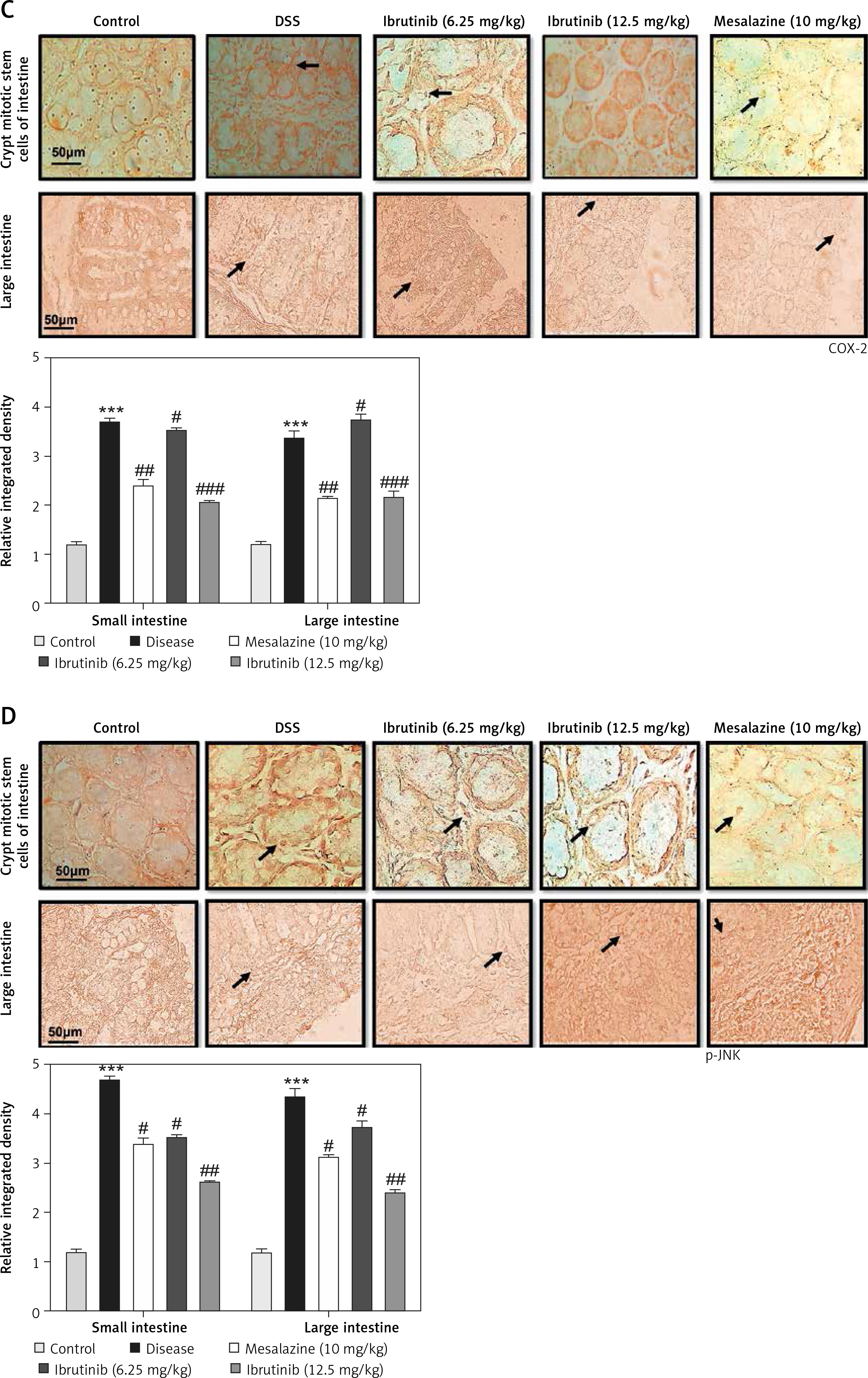
With slight modifications, immunohistochemical staining was performed as previously described. To retrieve the antigen, the slides were subjected to proteinase K treatment and then washed with PBS and the peroxidase activity was further quenched. Slides were blocked with normal goat serum (NGS) and incubated overnight in mouse anti-nuclear factor-κB (NF-κB), mouse anti-COX2 (dilution 1 : 100, Santa Cruz Biotechnology, United States), and mouse anti-nuclear p-JNK (dilution 1 : 100, Santa Cruz Biotechnology, United States). After washing, secondary antibodies (dilution 1 : 50) were applied in a humidified chamber, and then ABC reagent (SCBT, United States) was applied. After washing, slides were stained by DAB and coverslipped with a mounting medium. ImageJ software was used to quantitatively determine hyperactivated COX2, p-JNK, and p-NF-κB in the intestine/total area by optimizing the background of images according to the threshold intensity and analyzing COX2, p-JNK, and p-NF-κB positive cells at the same threshold intensity for all groups. The intensity is expressed as the relative integrated density of the samples relative to the control.
All the samples were homogenized using lysis buffer (1M Tris-HCl, 5 M NaCl, sodium deoxycholate 0.5%, sodium dodecyl sulfate 10%, sodium azide 1%, and NP-40 10%) further supplemented with a protease inhibitor, i.e. phenyl methyl sulfonyl fluoride (PMSF). After sonification and centrifugation, the protein concentration of the lysate was determined using a Bicinchoninic Acid kit (BCA) (Pierce, Rockford, IL, United States) following the guidelines of the manufacturer. An equal amount of protein (30 µg per sample) was electrophoresed on 10% SDS-PAGE gels, followed by transferring the protein to polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, United States). After blocking the PVDF membranes using skim milk to reduce binding of the nonspecific protein, the PVDF membrane was incubated at 4°C for the whole night with the primary antibodies COX2, p-NF-κB, TNF-α, p-Akt, p-PI3K, and p-GSK3β. β-actin was used as a loading control. The following day, membranes were incubated with secondary antibodies, and western bands were visualized on X-ray film using an ECL detection reagent following the instructions of the manufacturer (Amersham Pharmacia Biotech, Piscataway, NJ, United States). Image J software was used for the analysis of WB bands and was analyzed relative to β-actin.
Our laboratory protocols were adopted for these assays. Briefly, the glutathione (GSH) level [25] for measuring catalase and glutathione S-transferase (GST) activity was analyzed according to the procedure described by Rahman et al. (2020) [26].
The method mentioned previously was used to determine lipid peroxidation [27]. Mice intestinal tissue was homogenized by the aid of a polytron homogenizer in 20 mM Tris HCl. Following supernatant collection, ferrous ammonium sulphate was added to the clear supernatant incubated at 37°C to calculate lipid peroxidation. Later on, 2-thiobarbituric acid (TBA) was added to the above mixture and the absorbance was calculated at 532 nm by employing a microplate reader.
Morphological data were analyzed using ImageJ software (NIH Image J system, Bethesda, MD). The results were expressed as mean ± standard error of the mean (SEM, n = 5) and statistically analyzed via one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. Values of p < 0.05 were considered as statistically significant. GraphPad Prism (GraphPad, San Diego, CA, USA) was used for analysis of data.
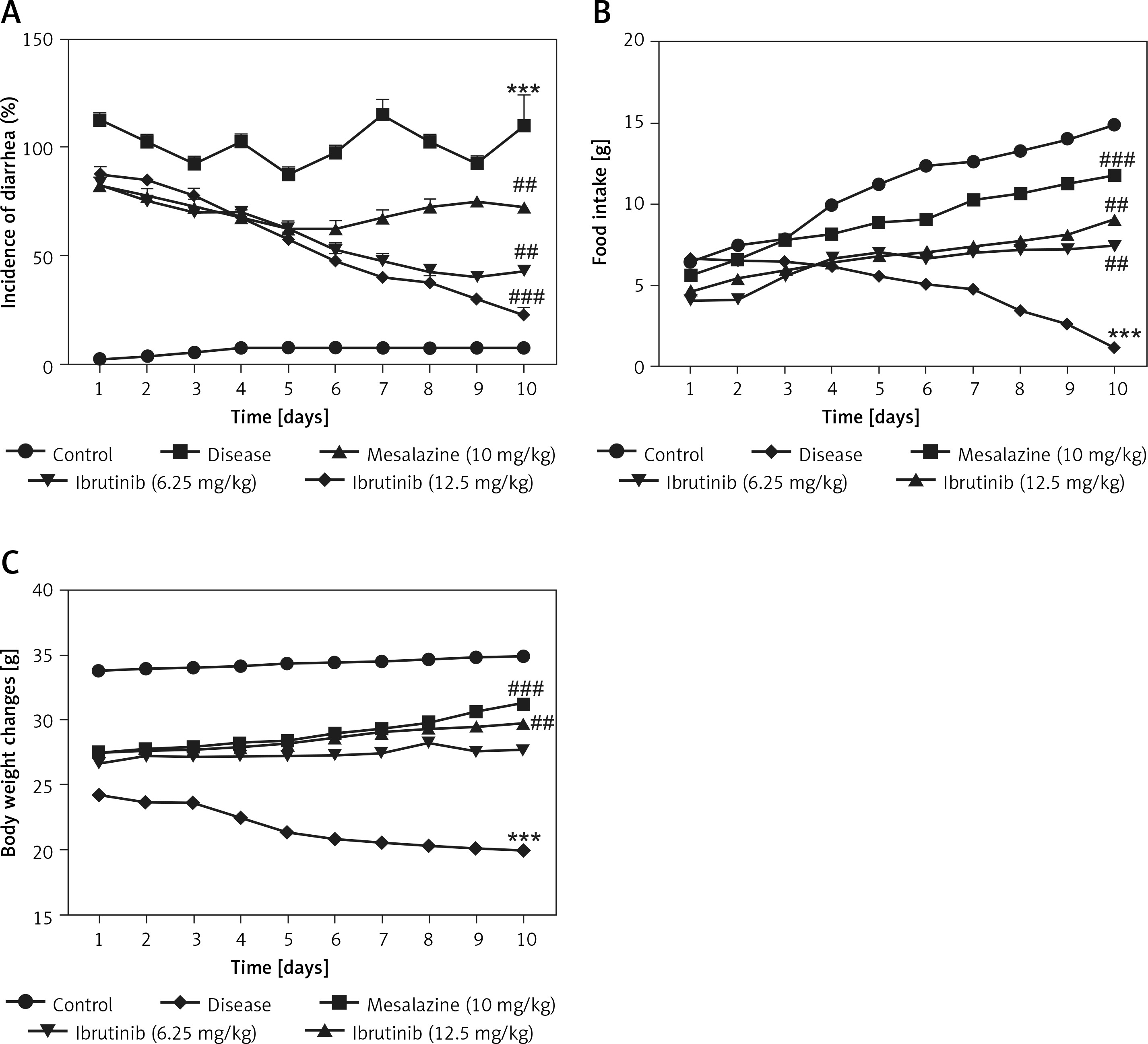
Mice in all 5 groups were examined for physical and pathological symptoms of IBD including weight loss, food intake, and diarrheal tendency. Mice in the saline group showed normal locomotion, eating, and drinking behavior with no signs of diarrhea or weight loss compared to the DSS group with a decline in physical assessment accompanied with bloody diarrhea (Figures 1 A–C). Mice treated with ibrutinib at a dose of 6.25 mg/kg improved reluctance to movements and food intake with increased weight and less watery stools (Figures 1 A–C). However, ibrutinib at a dose of 12.5 mg/kg significantly attenuated diarrhea and weight loss in DSS-induced IBD, suggesting its dose-dependent effect. Mesalazine (10 mg/kg) showed a marked effect on physical symptoms including weight loss, diarrhea, and movement as compared to the DSS group (Figures 1 A–C).
Figure 1
Ibrutinib attenuated physical manifestations of DSS-induced IBD in mice. A – Diarrhea score. B – Food intake. C – Body weight changes. Data show the percent incidence of diarrhea at different time intervals. The severity of diarrhea is evident from 72 h (day 3) to 240 h (day 10) after DSS administration. As compared to the DSS group, ibrutinib (6.25 mg and 12.5 mg) pretreatment reduces this severity from 120 h (day 5) to mild and moderate type. All the mice were monitored during the whole experiment for diarrhea score, which was based on stool consistency. The data are expressed as mean ± SEM and were analyzed using one-way ANOVA followed by Tukey’s multiple comparison test, and n = 14/group. The symbol * shows a difference relative to the saline group and # shows a difference relative to the DSS group. The data presented are relative to saline and the disease group. The symbols *** and ### represent significant difference values of p < 0.001, while the symbol ## represents p < 0.01 values for significant differences and # represents p < 0.05
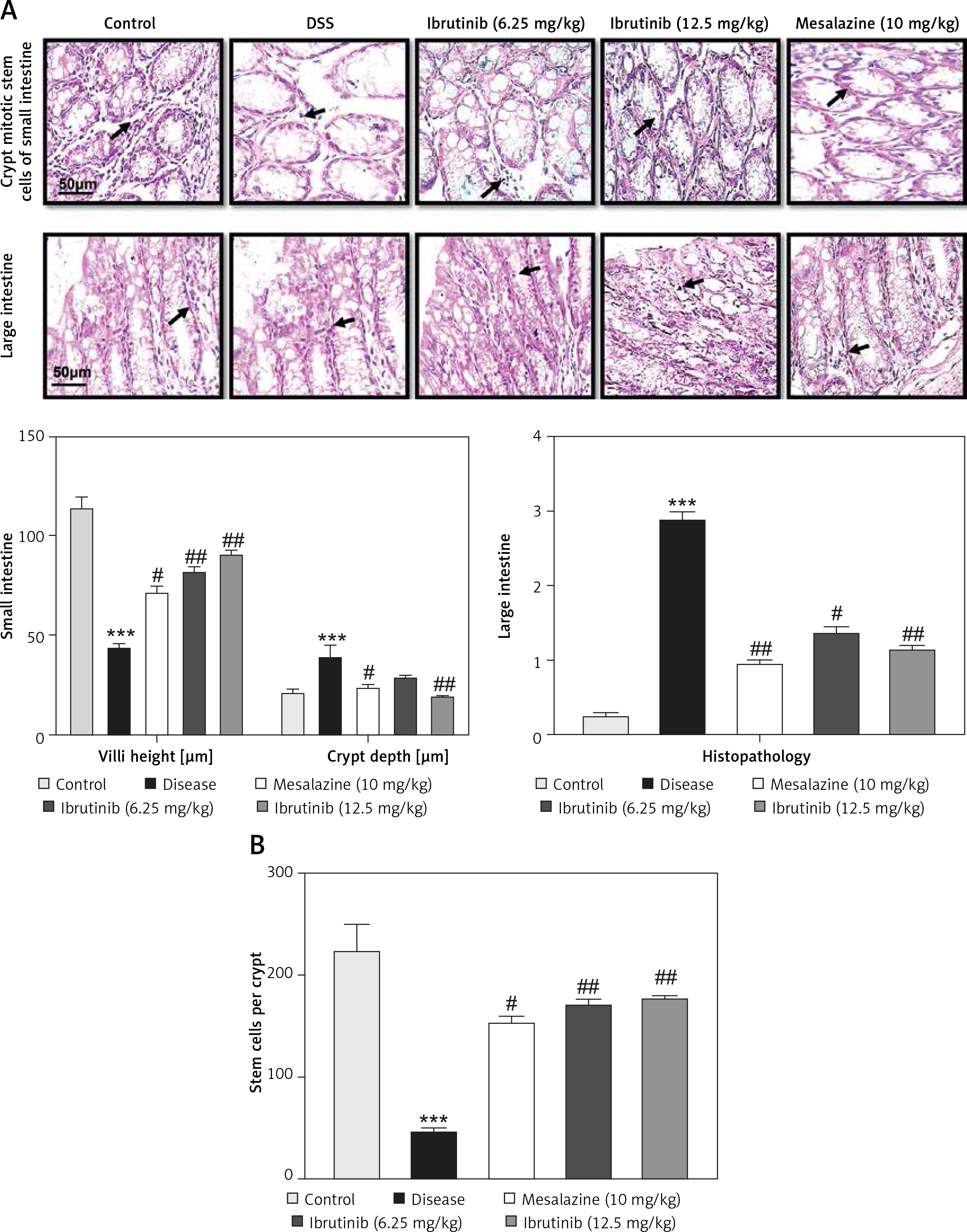
The saline group showed no pathological changes and normal architecture of both small and large intestinal tissues (Figure 2 A). In the DSS group, significant alterations were observed including epithelial destruction, inflammatory infiltration, and crypt damage, which are key indicators of colon tissue injury (Figures 2 A, B). In the 6.25 mg treated ibrutinib group, moderate inflammatory cells were noted, which can result in inflammation and edema formation (Figure 2). A significant protective effect was observed for ibrutinib at a dose of 12.5 mg, with no visualization of inflammatory cells, no evidence of crypt cell damage, further validated by no fissures on colon and intestinal cells, indicating normal architecture of colon tissues similarly to the control group. Some mild inflammatory cells were observed in the standard group (mesalazine 10 mg/kg) but no epithelial damage in extra- and intracellular spaces was found (Figures 2 A, B).
Figure 2
Effect of ibrutinib on histopathological injury induced by DSS in mice. A – Effect of ibrutinib on H&E staining for both small and large intestinal histological features in mice administered with DSS. B – Crypt mitotic stem cells of intestinal tissue. Photomicrographs were taken at 40×. magnification of both small and large intestinal jejunum across all groups. The saline group presents no histopathological changes with tall and intact villi, while the DSS treated group presents significant histopathological changes followed by severe epithelial atrophy of villi, enormous inflammatory cells infiltration within lamina propria, vacuolization, and edema both in mucosa and muscularis. The saline group presents a normal number of stem cells (arrow) in the crypt with no apoptotic index. The DSS group presents a complete loss of stem cells (arrow) in the crypt with an intense apoptotic index. The mesalazine group presents renewal of stem cells (arrow) in the crypt with suppressed apoptotic index, while ibrutinib administration causes the usual proliferation of stem cells (arrow) in the crypt with a reduced apoptotic index in a dose-dependent manner. The data are expressed as mean ± SEM and were analyzed using one-way ANOVA followed by Tukey’s multiple comparison test, and n = 7/group. The symbol * shows a difference relative to the saline group and # shows a difference relative to the DSS group. The data presented are relative to the saline and the disease group. The symbols *** and ### represent a significant difference values of p < 0.001, while the symbol ## represents p < 0.01 values for significant differences and # represents p < 0.05
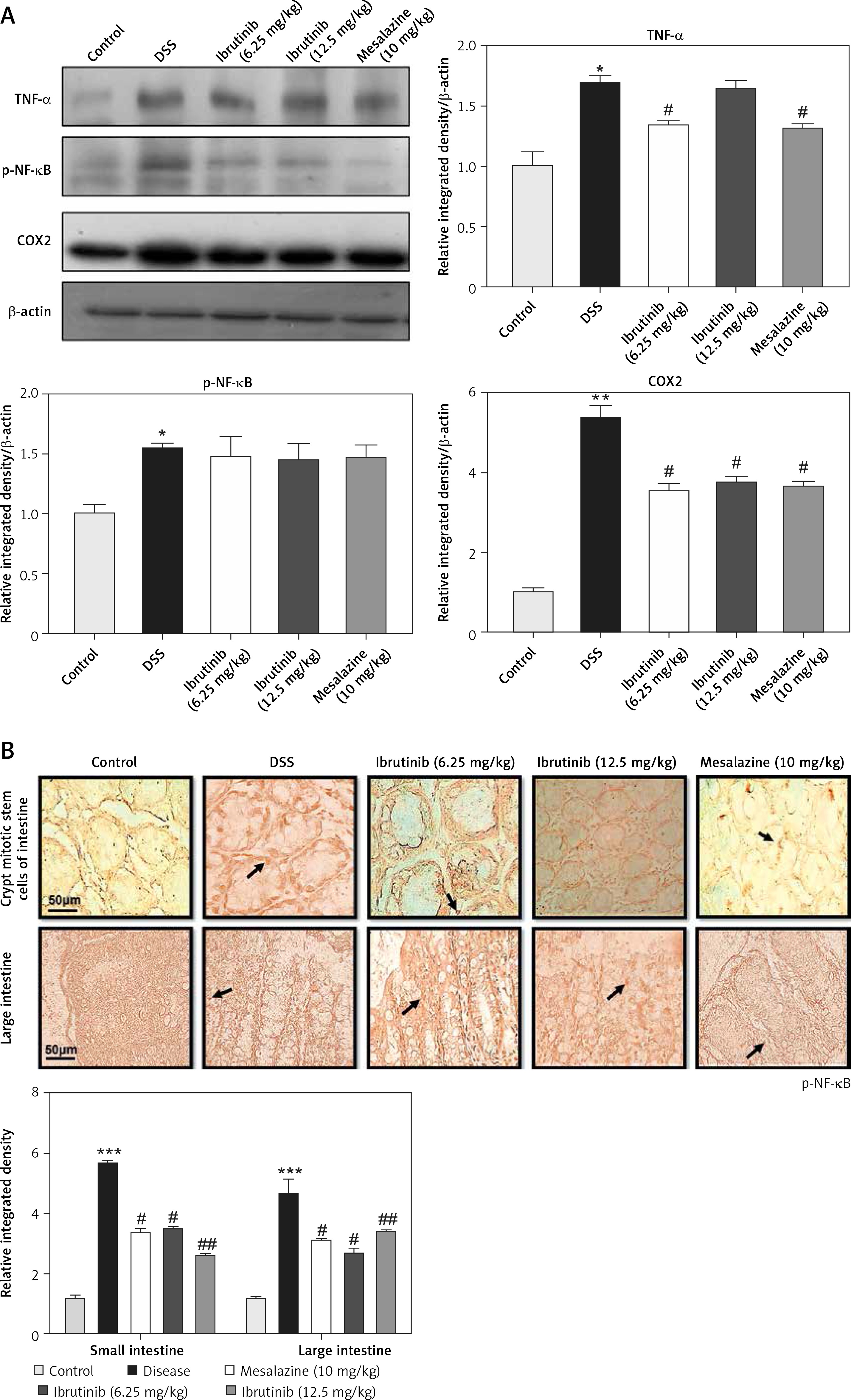
The expression of inflammatory mediators in intestinal cells was determined using immunohistochemistry and western blot for expression analysis of molecular markers of inflammation. Compared with baseline expression, the levels of TNF-α, p-NF-κB, and COX2 were significantly increased in the disease group as shown in Figure 3 A (*p < 0.05) in comparison to control. To further validate this, we performed immunohistochemistry and similar results were obtained (Figures 3 B, C). Treatment with ibrutinib significantly ameliorated the elevated inflammatory markers. Moreover, p-NF-κB is linked to downstream inflammatory cytokines; therefore we sought to determine whether ibrutinib could affect p-JNK level. An elevated level of P-JNK in the disease group was noted compared to that in the control group as shown in Figure 3 D while treatment with ibrutinib attenuated this elevated level of p-JNK relative to the disease group (Figure 3 D, # p < 0.05).
Figure 3
Ibrutinib attenuated IBD effect by targeting inflammation. A – Western blot analysis of TNF-α, p-NF-κB, and COX2. The data presented are relative to β-actin with n = 7 per group and the experiments performed twice. B – The presented images indicate the immunoreactivity of p-NF-κB The presented images indicate the immunoreactivity of COX-2 (C) and (D) p-JNK. Scale bar = 50 μm, magnification 40×. with n = 7/group. The data are presented as means ± SEM and were analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. The symbol * shows a difference relative to the saline group and # shows a difference relative to the DSS group. The symbols *** and ### represent significant difference values of p < 0.001, while the symbol ## represent values of p < 0.01 for significant differences and # represents p < 0.05. COX2 and JNK show cytoplasmic localization while p-NF-κB can be traced in the nucleus
Western blot analysis was performed to determine the effect of ibrutinib on the PI3K/Akt pathway (Figure 4). DSS reduced the expression level of these protective markers (*p < 0.05) while treatment with ibrutinib reversed the downexpression of these proteins and similar results were obtained as that of the standard (Figure 4, # p < 0.05).
Figure 4
Effects of ibrutinib on enzymatic and non-enzymatic oxidants. Effects of DSS and ibrutinib on levels of GSH (A); GST (B); CAT (C), and LPO (D). The data are presented as means ± SEM and were analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. The symbol * shows a difference relative to the saline group and # shows a difference relative to the DSS group. The symbols *** and ### represent significant difference values of p < 0.001, while the symbol ## represents p < 0.01 values for significant differences and # represents p < 0.05
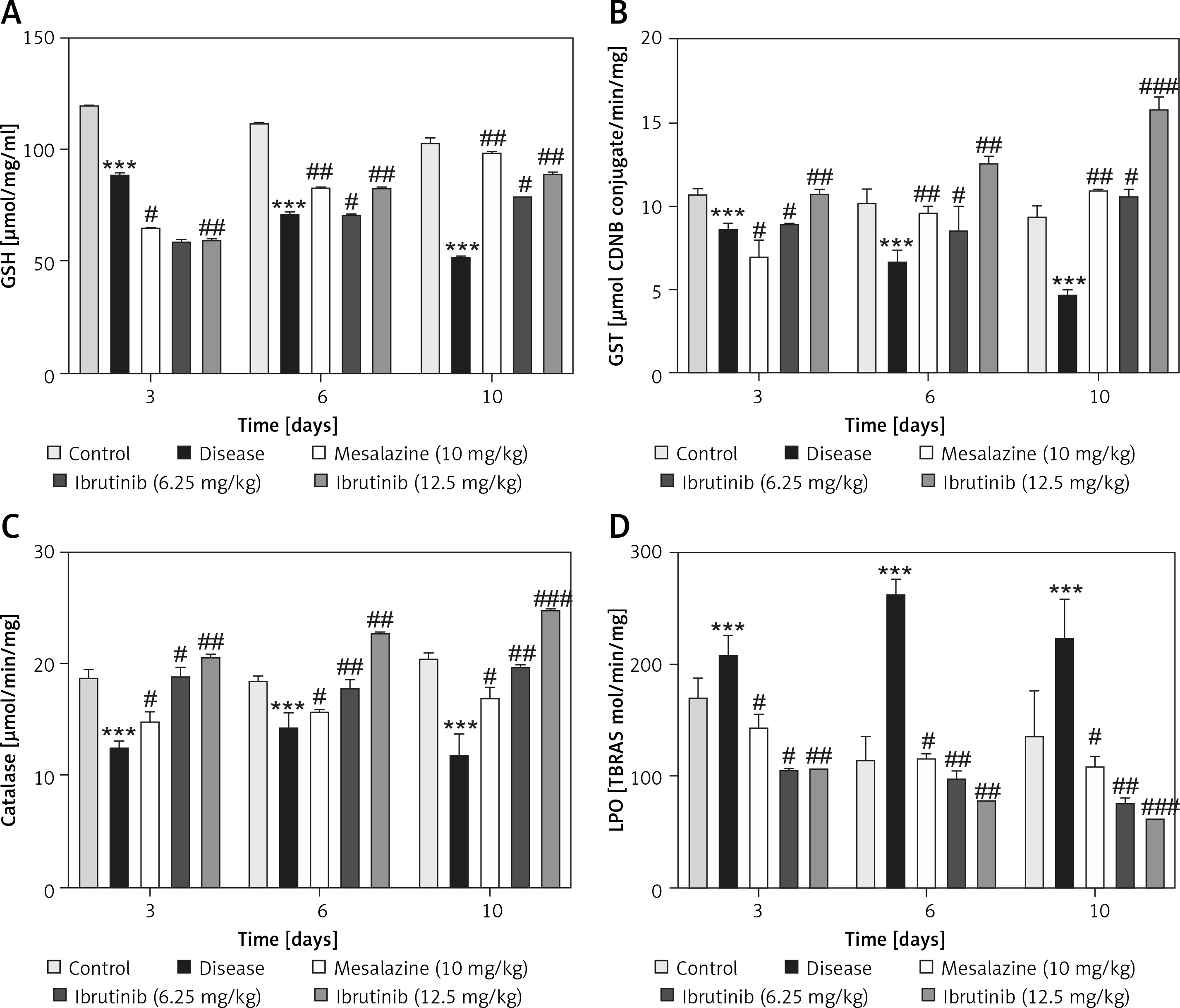
As first-line indicators of oxidative damage, the levels of enzymatic and non-enzymatic oxidants such as GST and GSH were measured in intestinal tissues on the 3rd, 6th, and 10th day. Administration of DSS induced reactive oxygen species (ROS) along with the exhaustion of GSH and GST in the intestinal tissue homogenates (Figure 4 A, B). Treatment with ibrutinib increased the levels of GSH and GST in the intestine homogenates. Further, treatment with ibrutinib restored the level of CAT in both homogenates (Figure 4 C). Oxidative stress and ROS generated in hyperuricemia induced the formation of several detrimental products, including malondialdehyde (MDA), which can be measured by TBARS. Therefore, we performed a lipid peroxidation (LPO) assay to assess the degree of damage induced by LPO products, which were increased relative to the saline group (Figure 4 D). Ibrutinib and mesalazine treatment attenuated LPO in the intestinal tissue (Figure 4 D).
DSS induces dramatic changes in levels of enzymatic enzymes such as ALP, ALT, AST, and TB. Levels of liver enzymes were examined on the 3rd, 6th and 10th days of treatment. Normal levels of liver enzymes were detected in the saline group as compared to the DSS group on the 3rd, 6th, and 10th day respectively (Table I). However, ibrutinib (6.25, 12.5 mg/kg) significantly attenuated the levels of ALP, ALT, ASP, and TB, showing a potent effect on liver enzyme levels (Table I). Mesalazine (10 mg/kg) showed an effect on liver enzymes by attenuating levels of liver enzymes (Table I).
Table I
Effect of ibrutinib on liver profile in DSS-induced IBD. Data show the effect of ibrutinib on liver enzymes: 3rd day, 6th day, and 10th day. The data are presented as means ± SEM and were analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. The symbol * show a difference relative to the saline group and # shows a difference relative to the DSS group. The symbols *** and ### represent significant difference values of p < 0.001, while the symbol ## represents p < 0.01 values for significant differences and # represents p < 0.05
In the current study, we investigated the protective effects of ibrutinib against DSS-induced IBD, which was mediated possibly by attenuating the oxidative and inflammatory cascade as ibrutinib exhibited anti-inflammatory potential in the previous studies [28]. Several studies supported the immunomodulatory role of ibrutinib and we further investigated the role of the favorable biological effects of ibrutinib, and we observed a dose-dependent response and the results are comparable to the standard group. Our study showed that ibrutinib treatment restored the physical parameters of colitis such as acute diarrhea, diminished water, and food intake, recovered the structural integrity of intestinal tissues, and normalized the endogenous nonenzymatic antioxidant protein. We observed here that ibrutinib mediated these effects possibly by attenuating the underlying inflammatory cascade such as TNF-α and NF-κB and by enhancing the protective mechanism (PI3K/Akt/GSK3-β).
The detrimental effects of DSS can be attributed to the dismantling of the basal epithelium, which can provoke thinning, atrophy, and necrosis of mucosa, further linked to crypt vanishing of intestinal goblet cells and infiltration of immune cells into the colon and intestine [29]. Such infiltration will provoke the release of inflammatory cytokines such as IL-1β, IL-6, and JNK, which exacerbated inflammatory reactions in intestinal tissues [30]. NF-κB and c-Jun have been reported to play a crucial role in inflammation caused in the colon and significant inhibition of these markers reduces the pathogenesis of IBD [31]. Moreover, these mediators are cross-linked to other inflammatory cytokines and chemokine levels and facilitate the migration of immune cells to the colon, which relates to the pathogenesis of human IBD [32]. To further reveal the relative role of the inflammatory markers and their associated cascade in our model, we demonstrated that these markers could exaggerate pathogenicity, as a substantial level of inflammatory cytokines was documented in the disease group and ibrutinib attenuated the level of both histological pathogenesis and inflammatory mediators. Administration with ibrutinib alleviated all the physical manifestations induced by the DSS administration, suggesting a preferable candidate that protects the mucosa. Furthermore, treatment with ibrutinib significantly prevented DSS-induced intestinal damage such as shortening of villi, the disappearance of mitotic crypt cells, increase of goblet cell emptying, excess of inflammatory cell infiltration and edema, which were confirmed by histopathological assessment. All these histopathological aspects reveal the mucoprotective effects of ibrutinib. Moreover, how ibrutinib effectively managed these targets needs to be further investigated.
Our results are in line with previous studies as DSS administration suppressed the levels of antioxidant mechanisms such as GSH, GST, and catalase, and increased the level of LPO [33]. The ibrutinib group showed a marked increase in the antioxidant enzymes, while the level of the MDA was attenuated promisingly. It was found that treatment with ibrutinib not only increases the level of the antioxidant enzyme by repressing ROS production but also reinforces its anti-oxidant potential through inhibition of lipid peroxidation. ROS production initiated a series of downstream signaling via the pivotal transcriptional factor NF-κB, which regulates several cellular activities such as inflammation, cell division, apoptosis, differentiation, and development. DSS can trigger activation of the transcriptional factor NF-κB in intestinal tissue and consequently up-regulate the expression of several genes, especially those concerned with the production of pro-inflammatory cytokines [34]. The excessive production of the proinflammatory cytokines via the positive feedback mechanism further activates NF-κB signaling and facilitates the swift discharge of mucins from the goblet cells, rendering them cavitated [35].
As repetitive clinical trials of different protectants that target a single step in different pharmacological disorders have failed previously, ibrutinib in this study ameliorated the detrimental effects of hyperuricemia by targeting multiple stages, such as inflammation, oxidative stress, and cellular degeneration. Our results demonstrated that a protectant acting at different stages may be more advantageous to intervene in the vicious cycles.
As an upstream activator of NF-κB, the phosphoinositide 3-kinase (PI3K)/AKT signaling pathway is implicated in multiple cellular processes, such as survival, proliferation, differentiation, and apoptosis, and the PI3K/Akt signaling pathway is linked to major degenerative diseases. PI3K activation also regulates RhoA kinase, a key regulator of actin cytoskeleton organization, and may influence both degeneration and protection depending on the signal strength. Our results showed attenuated expression of PI3K in the DSS, whereas ibrutinib treatment enhanced the PI3K/Akt signaling pathway, suggesting prosurvival signaling.
Conversely, ibrutinib exhibited a protective effect on liver enzymes as a relative safe profile as no impairment was observed in the liver, further assisted by serum ALT, AST, ALP, and TB levels. Prospectively, the present study was carried out on the comparison of study drug ibrutinib with the standard market drug mesalazine that is mostly used in IBD.
Overall ibrutinib has attractive potential because of the significant decrease in the hepatic enzyme level in blood accompanied by reduced inflammation in intestinal tissues. Similar results were noted in the present study, with all the effects alleviating the condition of the treatment group injected with 12.5 mg/kg body weight of ibrutinib.
In conclusion, the current study demonstrated that seven consecutive days of intraperitoneal administration of ibrutinib (6.25 mg/kg and 12.5 mg/kg) alleviated hyperuricemia in an animal rodent model, and produced significant anti-gout effects in a dose-dependent manner. In conclusion, IBD activates several proinflammatory mediators including p-NF-κB and is further linked to ROS generation. Ibrutinib attenuated hyperuricemia-induced oxidative stress and the inflammatory cascade, possibly by modulating the ROS/NF-κB pathway, potentially accounting for its protective effects against intestinal necrosis.