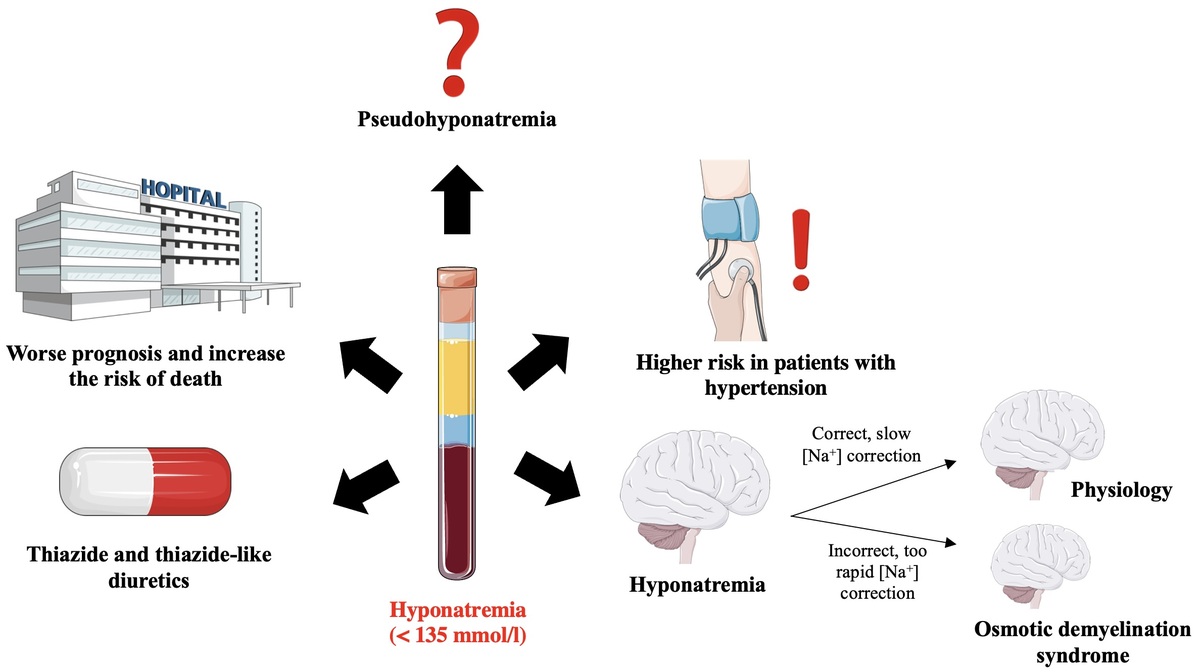
Physiology of sodium homeostasis in humans
The sodium content in an adult subject with 70 kg body weight is approximately 4200 mmol sodium (≈60 mmol/kg bw). Sodium is the main cation in the extracellular space, which contains 91% of the body sodium content. Plasma is a significant part of the extracellular space of the body. The physiological concentration of sodium in plasma is in the range between 135 and 145 mmol/l. Plasma sodium is rapidly exchangeable and its concentration depends on the current dietary sodium intake, renal water homeostasis and current volume of plasma, related to the water balance of the body. The intracellular space contains only 9% of the total sodium content in the body. In this space, the sodium concentration is 10–20 mmol/l (Figure 1). During recent years it has been documented that about 1/3 of the sodium content in the body (≈20 mmol/kg bw) is exchangeable to a small extent and is deposited in the bones and in glycosaminoglycans, located mainly in the subcutaneous tissue (Figure 1) [1–3].
Figure 1
The sodium homeostasis [1–3]. The content of sodium in the body was calculated for a human weighing 70 kg
Sodium intake has changed over the centuries. The paleolithic diet contained < 0.8 g sodium/day (< 2 g NaCl/day) [4]. Due to the fact that in the Paleolithic Age the availability of sodium in food (derived obviously only from unprocessed food products) was low, and in the course of evolution the human body developed numerous mechanisms aimed at precise regulation of natremia and enabling survival in an environment containing little amounts of sodium [5]. Along with the development of civilization, methods of food processing with the use of sodium chloride have been introduced. Therefore, sodium content in the human diet was increasing. Currently, in the United States and Europe the sodium content in the diet is over 5 g/day (about 13 g NaCl/day) [6], and industrially processed foods become the main source of sodium [7].
Sodium is absorbed primarily in the middle and distal parts of the small intestine by: (1) passive transport with an electrochemical gradient; (2) co-transport with nutrients such as glucose or amino acids; (3) co-transport with chloride anions; or (4) anti-transport – exchange with protons [8].
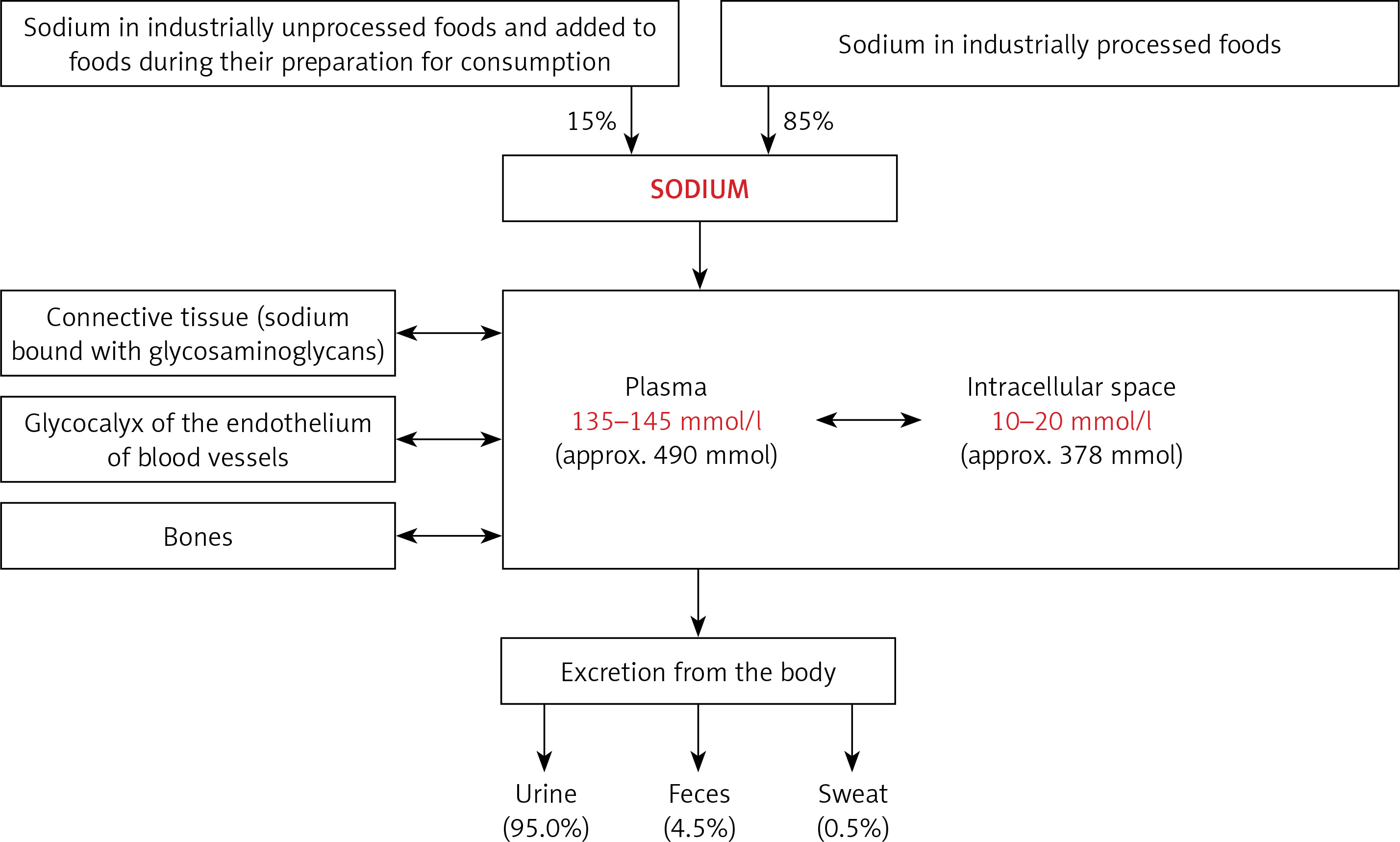
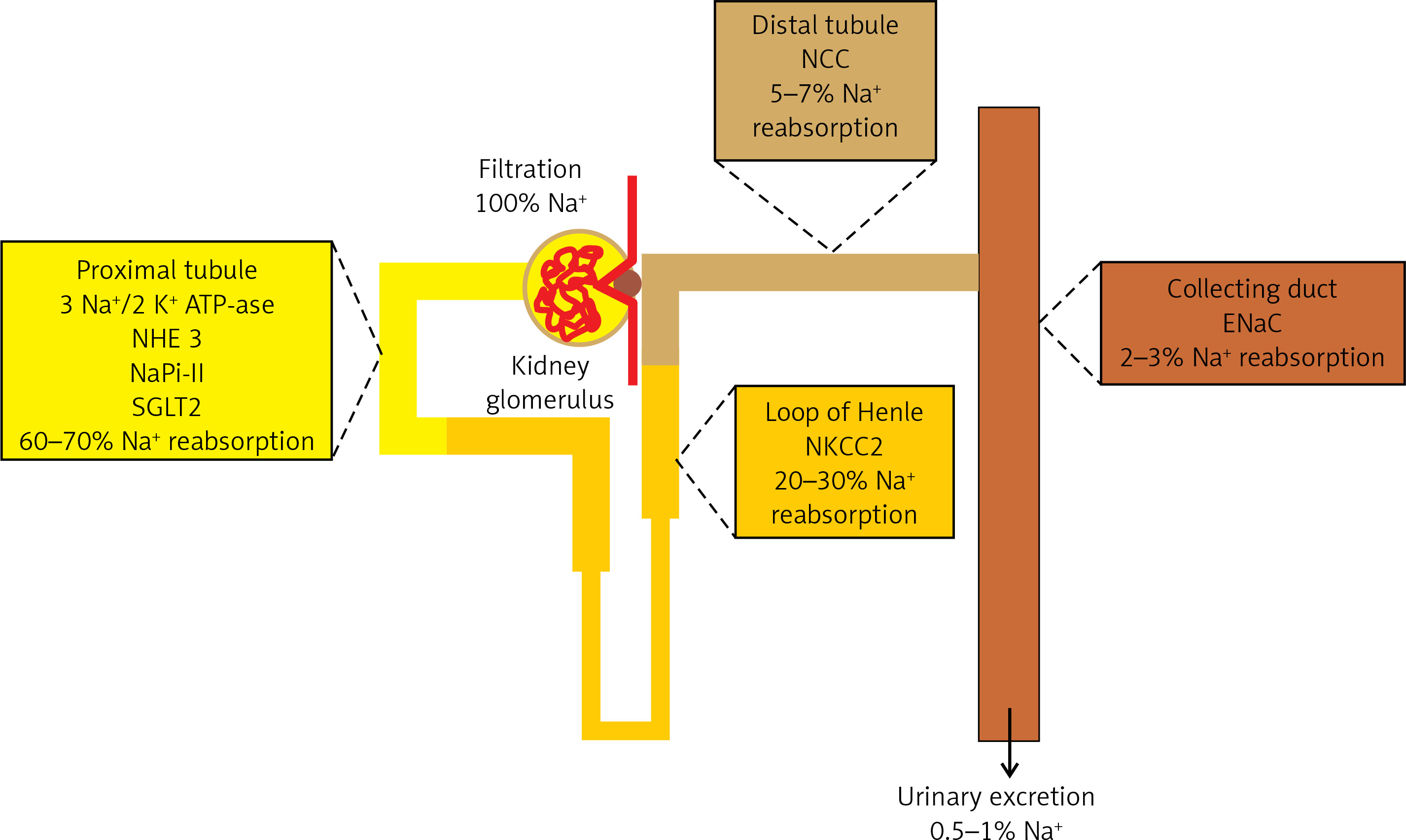
The kidneys play the most important role in the regulation of sodium homeostasis and in the physiological conditions 95% of ingested sodium is excreted through the kidneys, 4.5% in the faces and 0.5% through the skin (Figure 2). Maintaining natremia is regulated by a number of factors that affect the reabsorption of sodium in different parts of the nephron. Sodium reabsorption takes place along the almost entire length of the nephron, except for the descending limb of the loop of Henle (Figure 2) [5, 8, 9].
Figure 2
The role of the kidneys in the sodium homeostasis [6, 8, 9]. Sodium reabsorption takes place along almost the entire length of the nephron, except for the descending limb of the loop of Henle
NHE3 – sodium-hydrogen exchanger 3, NaPi-II – sodium-phosphate cotransporter, SGLT2 – sodium/glucose cotransporter 2, NKCC2 – Na-K-Cl cotransporter 2, NCC – sodium-chloride symporter, ENaC – epithelial sodium channel
In recent years, it has been shown that the interstitial water space (located mainly in the subcutaneous tissue) plays an important role in the regulation of sodium homeostasis. The role of this water space is mainly based on the osmotic inactivation of sodium by binding it to sulfate derivatives of glycosaminoglycans (Figure 1). Accumulated in the interstitial water space, sodium is osmotically inactive. Therefore, the accumulation of sodium in this water space does not cause simultaneous water retention. The mononuclear phagocyte system (MPS) macrophages stimulate the tonicity-responsive enhancer binding protein, vascular endothelial growth factor C (VEGF-C), vascular endothelial growth factor C receptor (VEGF-C-R), nitric oxide (NO), lymphangiogenesis and vasculogenesis pathways. These trails are involved in the storage or release of sodium from the interstitial water space. Increased sodium intake is a factor stimulating the above-mentioned metabolic pathways [3, 10–12].
The glycocalyx on the surface of vascular endothelial cells also plays an important role in the regulation of sodium metabolism (Figure 1). The glycocalyx of vascular endothelial cells is thought to act as a buffer to prevent excess sodium from the blood from reaching the tissues. It is estimated that the volume of glycocalyx in the body is about 1.5 l, which allows about 7 g of sodium to bind [13].
Another recently discovered regulating factor of sodium homeostasis is the phase of increased natriuresis regulated by an endogenous endocrine factor, in part related to the biological rhythms of aldosterone and cortisol secretion. Increased natriuresis episodes may have weekly, monthly or longer cycles and does not depend on the current body sodium homeostasis [10, 14].
The natremia, to a small extent, reflects the systemic sodium resources and mainly depends on the proportion of dissolved substances and water in the extracellular water space. The sodium cation hardly penetrates the cell membranes and, therefore, together with glucose and urea, determines the osmolality of body fluids. Cell membranes (with the exception of specialized cell membranes) are water permeable due to the constitutive expression of aquaporins (AQP; water channels). Therefore, dysnatremias are mainly the result of disturbances in the water management of the body. The reduction of osmolality (mainly determined by [Na+]) in the extracellular water space leads to the displacement of water from the extracellular to the intracellular space and to cell swelling, e.g. in the central nervous system (CNS) (mainly astrocytes that express AQP-4). Increasing the osmolality (mainly determined by [Na+]) in the extracellular water space leads to the displacement of water from the intracellular to the extracellular space and to a reduction in the cell volume [1, 15–17].
Hyponatremia: definition, classification and epidemiology
Hyponatremia is defined by a plasma sodium concentration lower than 135 mmol/l [16, 18, 19]. There are several classifications of hyponatremia, and therefore, due to the current concentration of sodium ions in the plasma, several degrees of severity of hyponatremia can be distinguished:
– Mild hyponatremia: plasma sodium concentration is between 130 and 134 mmol/l;
– Moderate hyponatremia: plasma sodium concentration is between 125 and 129 mmol/l;
– Profound hyponatremia: plasma sodium concentration is below 125 mmol/l.
The above-described classification of hyponatremia is to some extent arbitrary, however it is commonly used in the literature [1, 16, 18, 19]. As it is discussed below, in patients with moderate and profound hyponatremia, a clear mortality increase is observed [20, 21].
Due to the time criterion, hyponatremia is classified into:
– Acute hyponatremia when its documented duration is less than 48 h;
– Chronic hyponatremia when its duration is documented for at least 48 h.
When it is not possible to classify hyponatremia by duration, it should be considered as chronic.
A clinical classification of hyponatremia is related to the total body water status, i.e.:
– Hyponatremia with clinical signs of volume depletion, occurring in approximately 16% of patients with hyponatremia, is due to more electrolyte loss than water loss. The kidneys, for normovolemia, maintance increase the reabsorption of water, which leads to further dilution of the extracellular fluid and thus worsens the hyponatremia;
– Hyponatremia with normal clinical status of hydration, occurring in approximately 2% of patients with hyponatremia, is caused by disturbances in the excretion of the so-called free water, i.e., without dissolved electrolytes, via the kidneys;
– Hyponatremia with clinical signs of overhydration occurs in approximately 21% of patients with hyponatremia [1, 16, 19].
In each patient with a decreased plasma sodium concentration, the possibility of so-called pseudohyponatremia, which is only a laboratory abnormality, should be taken into account. Pseudohyponatremia occurs in patients with a low serum sodium concentration measured by routine methods that measure sodium concentration in whole serum and subsequently convert it to serum water sodium concentration (assuming that water is 93% of the plasma weight), when the following coexist: severe hypertriglyceridemia, severe hypercholesterolemia or severe hyperproteinemia (Figure 3).
Figure 3
Pseudohyponatremia. Pseudohyponatremia occurs in patients with low plasma sodium concentration measured by routine methods that measure sodium concentration in whole serum and subsequently convert it to serum water sodium concentration (assuming that water is 93% of the plasma weight) when the following coexists: severe hypertriglyceridemia, severe hypercholesterolemia or severe hyperproteinemia. In this laboratory anomaly, normal plasma osmolality, i.e. 275–295 mOsmol/kg H2O, was found. The results of direct measurement of sodium concentration in serum water using an ion-selective electrode (i.e. potentiometry method) are reliable in such clinical situations
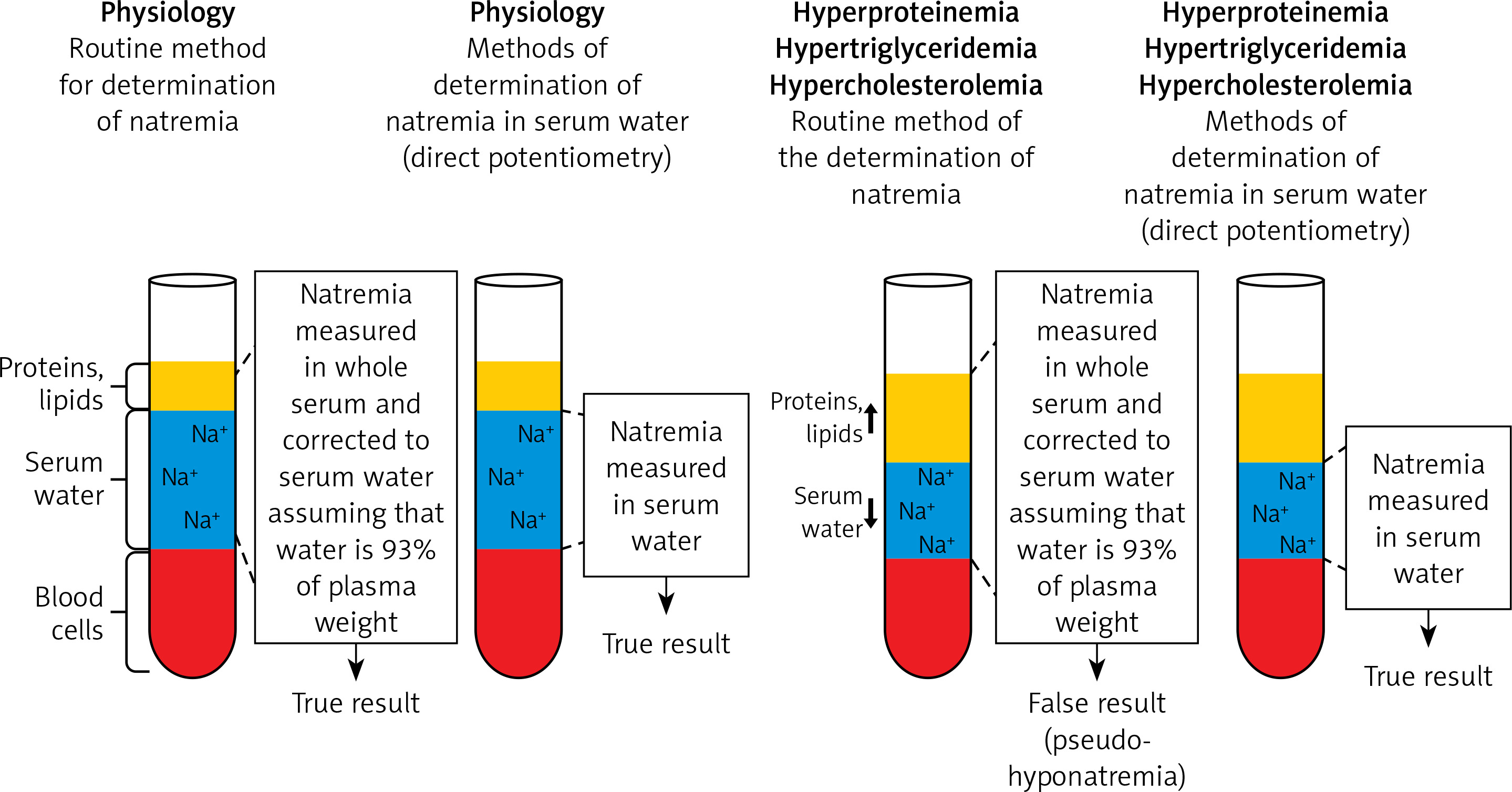
In this laboratory anomaly, normal plasma osmolality, i.e. 275–295 mOsmol/kg H2O is present. The results of direct measurement of sodium concentration in serum water using an ion-selective electrode (i.e. potentiometry method) are reliable in such clinical situations. Pseudohyponatremia obviously does not require treatment [22, 23].
Hyponatremia is a common electrolyte disturbance in clinical practice and its occurrence most often reflects an unfavorable prognosis. It is estimated that hyponatremia occurs in 5–29% of hospitalized patients [20, 24–26]. In a study by Zhang and Li, involving 17693 hospitalized older patients (aged 84.6 ±3.5 years), the prevalence of hyponatremia was 24.7% [21]. Mohan et al., in a study involving 14697 participants from the general population, showed that the incidence of hyponatremia was 1.7%. It occurred more often in women and its incidence increased with age of the participants. In patients with arterial hypertension, the incidence of hyponatremia was 2.9% and the risk of its occurrence was more than 1.5 times higher than in the general population (OR = 2.57; 95% CI: 1.77–3.73, p < 0.001) [27]. Results an of observational study suggest that hyponatremia is more common in the summer [28]. A study by Jönsson et al. in Sweden found that more cases of drug-induced hyponatremia were reported in the warmer season, with a peak in June [29].
In a meta-analysis of 81 studies by Corona et al., including 850222 participants, moderate hyponatremia increased the risk of death from any cause (RR (relative risk) = 2.60; 95% CI: 2.13–2.93) [30]. Zheng et al. showed that patients with moderate or profound hyponatremia were characterized by a higher risk of death compared to those with mild hyponatremia (moderate versus mild hyponatremia: OR (odds ratio) = 6.92; 95% CI: 2.53–18.92; profound versus mild hyponatremia: OR = 4.54; 95% CI: 1.05–9.58) [20]. A study by Zhang and Li showed that in older patients (aged 84.6 ±3.5 years), when compared with mild hyponatremia, moderate and profound hyponatremia were associated with significant increases in in-hospital mortality (OR = 1.89; 95% CI: 1.54–2.33 and OR = 2.66; 95% CI: 2.06–3.43, respectively; p < 0.01) [21].
In a meta-analysis published by Sun et al., including 7 observational studies involving 742979 participants, the influence of hyponatremia on the risk of death in patients with chronic kidney disease was assessed. It was shown that hyponatremia significantly increased the risk of death from any cause by as much as 34% (HR = 1.34; 95% CI: 1.15–1.57) [31]. In a meta-analysis of 15 clinical studies, including 10745 patients after stroke, Shima et al., found that hyponatremia increases the risk of death in the short (90 days) period after stroke (OR = 1.73; 95% CI: 1.24–2.42) [32]. Similar observations were made by Chen et al. in a meta-analysis of 12 clinical studies including 21973 participants. These authors found that hyponatremia increased the risk of death both in the short- and long-term in patients after stroke (RR = 1.61; 95% CI: 1.33–1.96 and RR = 1.77; 95% CI: 1.27–2.47, respectively) [33]. Moreover, in a meta-analysis of 20 clinical trials, completed by Ma et al., the influence of hyponatremia on the risk of short- and long-term mortality in patients after acute coronary syndrome was assessed. It was shown that hyponatremia significantly increased short- and long-term mortality (RR = 2.18; 95% CI: 1.96–2.42 and HR = 1.74; 95% CI: 1.56–1.942, respectively) [34].
Akbar et al. completed a meta-analysis of 8 clinical studies, including 11493 participants to assess the impact of hyponatremia on the prognosis of patients with coronavirus disease 2019 (COVID-19). It was shown that hyponatremia significantly increased the risk of worse prognosis (i.e. defined as increase of COVID-19 severity or prolonged hospitalization or death) in patients with COVID-19 (OR = 2.65; 95% CI: 1.89–3.72, p < 0.001) [35]. In the Pattharwin study of 5712 patients with COVID-19, it was found that moderate/severe hyponatremia and mild hyponatremia during hospitalization, compared to normal serum sodium concentration, significantly increased the risk of acute kidney injury (by 386% and 88%, respectively), the need for mechanical ventilation (by 613% and 188%, respectively) and in-hospital mortality (by 292% and 55%, respectively) [36].
Hyponatremia: symptoms and complications
In the case of a reduction in sodium concentration in the extracellular water space, water is displaced from this space into the intracellular water space, which leads to cell swelling. In most tissues and organs, this phenomenon has no significant clinical consequences. The exception is the brain, located in the unstretchable skull. Swelling of cells of the CNS and the related increase in brain volume lead to mechanical injury to the CNS, determining the clinical symptomatology of hyponatremia [16, 17].
The symptoms of hyponatremia depend on the actual plasma sodium concentration and its duration. In mild and moderate hyponatremia, impaired perception and motor coordination may occur, which lead to an increased risk of falls [16, 17, 37]. A meta-analysis by Corona et al., including 15 clinical studies with 51879 participants, assessed the effect of hyponatremia on the risk of falls. It was shown that hyponatremia significantly increased the risk of falls by 114% (OR = 2.14; 95% CI: 1.71–2.67). Moreover, it was found that hyponatremia increased the risk of fractures, especially of the femoral neck, by 100% (OR = 2.00; 95% CI: 1.43–2.81) [38]. A meta-analysis of 26 clinical studies by Murthy et al. showed that hyponatremia increased the risk of fractures at all sites by 134% (OR = 2.34; 95% CI: 1.86–2.96) [39].
In the case of profound hyponatremia, apart from disturbances in motor perception and coordination, nausea and vomiting, confusion, headache, abnormal sleepiness, convulsions and coma may occur [16, 17, 37]. Hyponatremia in most cases does not cause ECG abnormalities [40, 41]. However, some ECG changes may occur in profound hyponatremia. Profound hyponatremia has been found to increase the risk of atrial fibrillation, atrioventricular block, sinus arrest, and peaked T waves [40].
Taking into account the above-mentioned symptoms of hyponatremia, the risk of complications and death, as well as the fact that the disorder may recur [42], it is important to thoroughly understand the pathogenesis of hyponatremia. This will enable effective treatment and prevention of hyponatremia relapse.
Hyponatremia: pathogenesis and diagnosis
From the pathophysiological point of view, the causes of hyponatremia are classified according to plasma osmolality and the patient’s hydration status [43]. Table I summarizes the most common clinical situations that predispose to hyponatremia, while Figure 4 shows a diagram of the diagnostic procedure depending on the plasma osmolality and the patient’s hydration status.
Table I
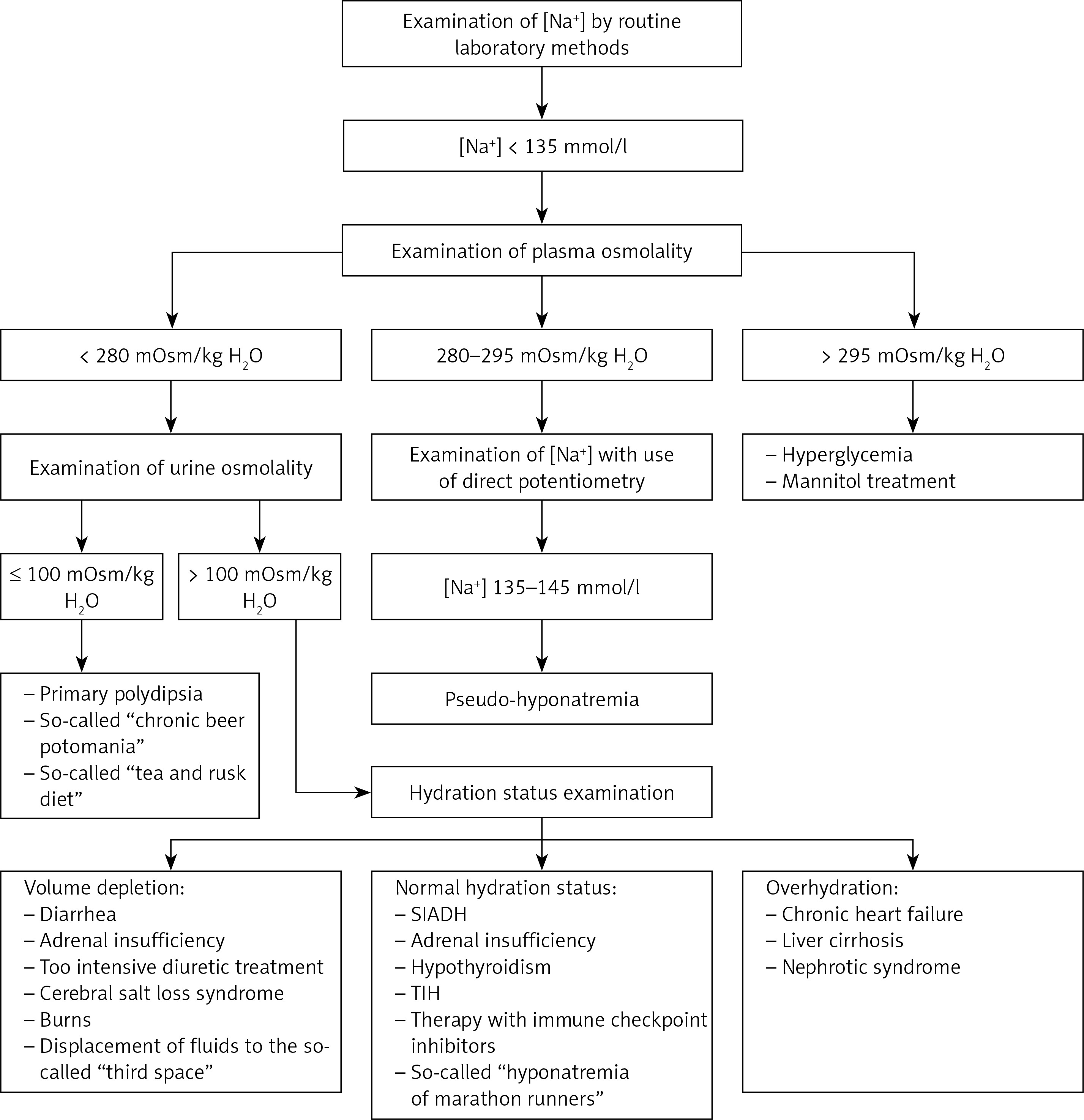
The incidence of various forms of hyponatremia was assessed by Fenske et al. in a study of 121 patients admitted to the hospital with a serum sodium concentration < 130 mmol/l. It was shown that in 35%, hyponatremia was due to a syndrome of inappropriate antidiuretic hormone hypersecretion (SIADH), in 32% by hyponatremia with signs of volume depletion, and in 20% by hyponatremia with signs of overhydration. Hyponatremia was diagnosed in 7% caused by thiazides or thiazide-induced hyponatremia (TIH), in 4% caused by primary polydipsia and in 2% caused by adrenal insufficiency [44].
Hyponatremia with plasma hyperosmolality
The coexistence of hyponatremia and plasma osmolality greater than 295 mOsm/kg H2O results from the displacement of water into the plasma and may occur in patients with severe hyperglycemia or after the use of mannitol. In patients with blood glucose concentrations ≤ 400 mg/dl, natremia is reduced by 1.6 mmol/l for every 100 mg/dl increase in blood glucose concentration. In patients with blood glucose concentration > 400 mg/dl, natremia decreases by 2.4 mmol/l with every 100 mg/dl increase in blood glucose concentration [1, 16, 19]. Treatment of hyponatremia with plasma hyperosmolality consists of reduction of the plasma osmolality, by means of hypoglycemic treatment in the case of hyperglycemia [1, 16, 19].
Hyponatremia with plasma hypoosmolality
Hyponatremia associated with plasma osmolality of less than 280 mOsm/kg H2O may be due to plasma dilution only or may be true hyponatremia, not solely due to plasma dilution.
In the first case, there is hyponatremia and urine osmolality ≤ 100 mOsm/kg H2O. This form of hyponatremia may occur in patients with psychogenic polydipsia (in a diet typical for Europe and the United States, water intake greater than 16 l/day may lead to hyponatremia). Hyponatremia resulting from psychogenic polydipsia occurs in 7% of patients with schizophrenia [45]. Hyponatremia resulting from plasma dilution only may also occur in subjects who undergo chronic so-called beer potomania. The following factors contribute to the pathogenesis of beer potomania hyponatremia: malnutrition (a diet low in salt and protein), cachexia and large amounts of alcohol consumed. These factors lead to a reduction in proteolysis, which results in a reduction in the amount of dissolved substances in the plasma, i.e. decrease in plasma osmolality. Reducing the amount of solutes reaching the kidneys (i.e., reducing the osmotic charge) reduces water excretion and, consequently, hyponatremia resulting from plasma dilution only [46]. Hyponatremia resulting from plasma dilution only may also occur in subjects who are chronically on a diet with a low content of electrolytes (the so-called tea-rusk diet) [16, 19].
In the second case, true hyponatremia not resulting from plasma dilution only, urine osmolality greater than 100 mOsm/kg H2O is observed. In these patients, the hydration status should be assessed in clinical examination. True hyponatremia, not only due to plasma dilution, as mentioned above, may occur with decreased, normal or increased water content in the body, assessed in clinical examination of the subject [22]. The causes of true hyponatremia not due to plasma dilution are summarized in Table I.
In hyponatremia with volume depletion, electrolyte loss is proportionally greater than water loss. Natriuresis is less than 30 mmol/l when sodium loss is not caused by its excessive excretion in the urine and the patients are not using diuretics [1, 16, 19].
Hyponatremia with normal hydration is caused by the impaired excretion of the so-called free (i.e. without dissolved electrolytes) water through the kidneys. An increased water content in the organism is observed with the simultaneous normal sodium content in the organism [1, 22]. In the diagnosis of hyponatremia with adequate hydration, hypothyroidism (hyponatremia occurs in about 4–14% of patients with hypothyroidism) and adrenal insufficiency should be excluded [19, 47–49]. Thereafter, a diagnosis for SIADH should also be completed. SIADH diagnostic criteria include: effective plasma osmolality less than 275 mOsm/kg H2O, urine osmolality greater than 100 mOsm/kg H2O, normovolemia in a clinical examination, urine sodium concentration greater than 30 mmol/l with adequate intake of sodium and water in the diet, no adrenal insufficiency, hypothyroidism and pituitary insufficiency, and kidney failure, and no recent use of thiazide or thiazide-like diuretics [16, 50]. The causes of SIADH are presented in Table II. Another cause of hyponatremia with normal hydration may be the so-called “marathon runner hyponatremia” due to the stimulation of vasopressin secretion through exercise and pain, and the intake of large volumes of fluids with little sodium, i.e. the so-called sports drinks during intense physical exertion [19, 22].
Table II
Causes of SIADH. Based on [16]
The use of diuretics significantly affects sodium homeostasis. Acetazolamide is an inhibitor of carbonic anhydrase in the proximal tubule of the nephron. The effect of acetazolamide is to reduce the production of hydrogen and bicarbonate ions, which reduces the sodium-hydrogen exchange. This results in an increased excretion of water, sodium and bicarbonate ions. Acetazolamide leads to an increase of sodium concentration in the distal tubule fluid, which intensifies the Na+/K+ exchange. This may result in hypokalemia (about 5% of patients using acetazolamide at a dose of 1.25–4 g/day). It should be emphasized that the diuretic effect of acetazolamide is short term. After 3 days of use, its diuretic properties disappear due to the appearance of compensatory mechanisms independent of carbonic anhydrase activity [51, 52]. In the proximal tubule, sodium/glucose cotransporter 2 (SGLT-2) also acts. SGLT-2 is a co-transporter involved in the reabsorption of glucose (80–90% glucose filtered by the kidney glomerulus) and sodium in the proximal tubule [53]. SGLT2 inhibitors stimulate natriuresis [54, 55]. It seems, however, that the influence of these drugs on the plasma sodium concentration was nonsignificant (weighted mean difference (WMD) = 0.15; 95% CI: –0.03 to 0.33) [56]. Lack of natremia reduction after SGLT2 inhibitors might be related to the fact that glucosuria leads to osmotic diuresis and consequently increased excretion of free-water. Moreover, SGLT2 inhibitors lead to sodium content reduction in the subcutaneous tissue [57, 58]. Results of the latter study might suggest that SGLT2 inhibitors interfere with osmotically inactive sodium storage. Moreover, results of the interventional study in patients with SIADH, showed that SGLT2 inhibitor empagliflozin improved hyponatremia correction [59]. Loop diuretics reduce the activity of Na+-K+-2Cl- symporter (NKCC2) in the thick ascending limb of the loop of Henle [60]. Results of the clinical study suggested that loop diuretics might not increase hyponatremia risk [61]. Mineralocorticoid receptor antagonists (MRA) and epithelial sodium channel (ENaC) blockers are active in the nephron collecting duct. These drugs stimulate natriuresis by reducing the activity of ENaC, which is involved in the reabsorption of sodium ions at the collecting ducts [62, 63]. The incidence of hyponatremia varies with the use of MRA. The RALES study showed no significant differences between spironolactone (25 mg/day) and placebo in serum sodium concentration [64]. Another study found that hyponatremia occurred in 3% of patients treated with spironolactone [65]. On the other hand, in the Eplerenone Post-AMI Heart Failure Efficacy and Survival Study (EPHESUS), it was shown that patients treated with eplerenone were characterized by a higher incidence of hyponatremia compared to placebo (15% vs. 11%, p = 0.0001) [66]. Finerenone is also more likely to induce hyponatremia compared to placebo (1.4% vs. 0.7%) [67]. In the case of ENaC blockers, there is also a certain risk of hyponatremia [68]. An interesting study by Mannheimer et al. involving 11213 patients assessed the effect of non-thiazide diuretics on the risk of hospitalization due to hyponatremia. It was shown that the use of furosemide (loop diuretic) did not significantly increase the risk of hospitalization due to hyponatremia, both during initiation of therapy (OR = 1.01; 95% CI: 0.81–1.24) and during chronic use (OR = 0.49; 95% CI: 0.44–0.54). The use of amiloride (ENaC blocker) increased the risk of hospitalization due to hyponatremia during initiation of therapy (OR = 6.21; 95% CI: 1.87–22.05), but not during chronic use (OR = 1.04; 95% CI: 0.66–1.57). Spironolactone (a mineralocorticoid receptor antagonist) increased this risk both short-tem after initiation of therapy (OR = 3.74; 95% CI: 2.80–5.01) and during chronic use (OR = 1.92; 95% CI: 1.68–2.19) [69]. Another particularly important cause of hyponatremia with normal hydration in hypertensive patients is thiazide-induced hyponatremia (TIH) [17]. This clinically important form of hyponatremia is discussed in detail in the next section of this paper.
It is worth mentioning that hyponatremia occurs in patients treated with modern anticancer drugs. In a study by Gupta et al., involving 78 patients treated with chimeric antigen receptor T-cell (CAR-T) therapy, the frequency of electrolyte disturbances was assessed. It was found that 30 days after the initiation of CAR-T therapy, hyponatremia was present in 51% of patients [70]. Moreover, hyponatremia may occur in up to 62% of patients treated with immune checkpoint inhibitors (ICI) [71]. In the case of CAR-T, the pathogenesis of hyponatremia includes the increased secretion of vasopressin due to the cytokine release syndrome, and especially due to the effect of interleukin 6 [68]. In turn, ICI-induced hyponatremia is caused by secondary autoimmune endocrinopathies such as anterior pituitary dysfunction, hypothyroidism or primary adrenal insufficiency [72].
In the case of hyponatremia with overhydration, the cause is an excessive intake of hypotonic fluids and/or impaired excretion of the so-called free (i.e. without solute electrolytes) water through the kidneys. An increase in the content of water and sodium in the body is observed [16, 19].
Management of hypertension and risk of hyponatremia
When analyzing the causes of hyponatremia in patients with arterial hypertension, the influence of lifestyle modifications (i.e. increasing physical activity, limiting salt intake (< 5 g/day), quitting smoking, reducing body weight, increasing the consumption of vegetables and fruits, and limiting alcohol consumption) and antihypertensive drugs on sodium balance should be discussed [73].
Increasing physical activity in accordance with the European Society of Hypertension and European Society of Cardiology (ESH/ESC) 2018 recommendations (e.g., at least 30 min of moderate dynamic exercise on 5–7 days per week) [73] is not likely to be the cause of exercise-related hyponatremia (EAH). As Rosner and Kirven point out, severe and life-threatening hyponatremia can occur during very intense physical exertion, especially during marathons (42.2 km), triathlons (8.3 km of swimming, 180 km of cycling, and 42.2 km of running) and ultramarathons (100 km) [74]. Most often, EAH is found in ironman triathlons and ultra-marathons [75]. It should be noted, however, that even in cases of heavy physical exertion, the incidence of EAH varies and is within the range of 0–51% [76]. This discrepancy highlights the tremendous complexity of both athletes’ environment and education, which have profound implications for both proper and inappropriate food and fluid intake strategies [77]. Life-threatening EAH is rarely reported [77]. A factor that increases the risk of EAH is fluid overload due to hypotonic fluids use, such as water or manufactured so-called sports drinks [78].
Dietary salt restriction may be associated with an increased incidence of hyponatremia in patients with hypertension. Therefore, it would also be advisable to measure plasma sodium concentrations before a low salt diet is recommended [79]. It is especially important in elderly hypertensive individuals. Bizimana conducted a case-control study involving 245 participants from Rwanda, where overall salt intake is low. It has been shown that in patients with arterial hypertension, a low-salt diet may significantly increase the risk of hyponatremia [80]. It should be emphasized that the number of published studies analyzing the impact of dietary salt restriction on the risk of hyponatremia in patients with arterial hypertension is very low.
Cigarette smoking might reduce plasma sodium concentration. Husain et al. showed that cigarette smoking increased the plasma vasopressin concentration in 10 out of 14 healthy volunteers [81]. Moreover, there are reports in the literature about the effect of nicotine replacement therapy on the risk of hyponatremia. In the description of the clinical case of a 39-year-old male treated with nicotine replacement therapy Finch et al. indicate that the constant concentration of nicotine in the serum delivered by the patch could induce hyponatremia through the continuous stimulation of vasopressin in this patient [82]. An experimental study by Yu et al. in Sprague Dawley rats showed that chronic nicotine exposure increased the expression of the vasopressin gene [83]. Moreover, it seems that the impact of nicotine replacement therapy on the risk of hyponatremia is low. A similar situation applies to the potential effects of drugs used in the treatment of cigarette addiction. Isolated cases of varenicline-induced hyponatremia have been reported in patients with schizophrenia [84].
Losing weight does not appear to influence the risk of hyponatremia. In a study by Bjørke-Monsen et al., including 119 obese patients, the effect of weight loss as a result of bariatric surgery on serum sodium concentrations was assessed. The slight increase in natremia was observed in patients with a significant weight loss (138.3 ±2.4 mmol/l vs. 141.8 ±1.9 mmol/l, p = 0.007). Twelve months after this procedure, serum sodium concentration was significantly higher in patients with normal body weight compared to overweight patients [85]. Thus, weight loss is not associated with the risk of hyponatremia.
Excessive alcohol intake may lead to hyponatremia. Therefore, reduction of alcohol consumption seems to reduce the risk of hyponatremia.
In conclusion, the lifestyle modification recommended to patients with arterial hypertension by the ESC/ESH does not significantly affect the risk of hyponatremia. The influence of diuretics on hyponatremia prevalence was already discussed. The thiazide or thiazide-like diuretic-induced hyponatremia is presented in the later part of this review paper. Results of some studies suggest that other antihypertensive drugs might also increase the risk of hyponatremia.
In a case-control population study by Falhammar et al., involving 11213 patients hospitalized due to hyponatremia, the impact of various antihypertensive drugs on the risk of this disorder was analyzed. It has been shown that the initiation of treatment with drugs such as calcium channel antagonists (OR = 1.86; 95% CI: 1.54–2.23), β-blockers (OR = 1.64; 95% CI: 1.35–1.96), angiotensin-converting enzyme inhibitors (ACEI) (OR = 2.24; 95% CI: 1.87–2.68) and angiotensin receptor blockers (ARB) (OR = 2.14; 95% CI: 1.72–2.67) was significantly associated with the risk of hyponatremia. Interestingly, for all these groups of drugs, their chronic use was not significantly associated with the risk of hyponatremia [86]. Different results in the case of chronic use of certain drugs and hyponatremia were obtained by Grattagliano et al. in a study involving 5635 patients aged > 65 years. It was shown that diuretics (p < 0.001), ACEI/ARB (p = 0.015) were significantly associated with the risk of hyponatremia [87]. The relationship between the use of ACEI and ARBs and the risk of hyponatremia was also shown in a case series (220 cases) by Yawar et al. Medicines accounted for 30% of cases of hyponatremia, of which ACEI and ARB were at the top of the list [88]. This preliminary observation indicates an increased risk of hyponatremia after initiation of therapy with ACEI and ARBs and its needs confirmation in further studies. The underlying mechanism of hyponatremia induced by above-mentioned antihypertensive drugs is unknown.
In summary, there are reports in the literature indicating that some other antihypertensive drugs may increase the risk of hyponatremia.
Thiazide or thiazide-like diuretic-induced hyponatremia
Hyponatremia caused by the use of thiazide or thiazide-like diuretics (TIHs) was first described 35 years ago [89]. TIH is clinically normovolemic hyponatremia (i.e. these patients do not have clinical signs of fluid overload or volume depletion) and is an important complication of thiazide or thiazide-related diuretics therapy. It should be noted that TIH is not the only cause of hyponatremia that may occur in patients using thiazide or thiazide-like diuretics [25]. The results of the clinical studies presented below suggest that hyponatremia is frequently observed in patients treated with these drugs.
TIH: epidemiology and risk factors
The frequency of TIH has not yet been precisely defined. In a population study completed among patients treated by general practitioners in Great Britain, the incidence of hyponatremia was determined in patients using thiazide or thiazide-like diuretics. The study included 950 patients treated with these drugs and it was found that 13.7% of them developed hyponatremia. Moreover, it was found that the incidence of hyponatremia increased with age of the studied patients [90]. In a clinical trial SHEP (Systolic Hypertension in the Elderly Program), hyponatremia was found in 4.1% of patients treated with chlorthalidone (at doses of 12.5 or 25 mg/day) [91]. In a retrospective study (follow-up, up to 10 years) assessing the risk of hyponatremia in patients with arterial hypertension, including 220 patients using thiazide diuretics and 2393 subjects not using them, hyponatremia was found in 30% of patients using these diuretics. Thiazide diuretic therapy increased the risk of hyponatremia by 61% (RR = 1.61; 95% CI: 1.15–2.25) [92]. The incidence of hyponatremia increases with age in patients using thiazide or thiazide-like diuretics. In the population study completed in Rotterdam, which included 5179 participants, it was found that among subjects aged 55-64 years, hyponatremia occurred in 6.1%, while in those aged 65–74 in 5.9%, and among those aged over 75 years of age in 11.6% of them. This study also found that the risk of hyponatremia in patients treated with thiazide or thiazide-like diuretics was significantly higher compared to patients not using these drugs (RR = 4.95; 95% CI: 4.12–5.96) [93].
TIH was probably the cause of hyponatremia in some of the patients participating in the above-described epidemiological studies, who developed hyponatremia during the use of thiazide or thiazide-like diuretics.
It is clinically important to establish whether there is a difference in the risk of developing TIH with the use of a different thiazide or thiazide-like diuretic. A meta-analysis of 12 clinical trials (5 studies with hydrochlorothiazide vs. indapamide and 7 studies with hydrochlorothiazide versus chlorthalidone) by Liang et al. showed no difference in the risk of hyponatremia with thiazide versus thiazide-like diuretics (mean difference: –0.14; 95% CI: –0.57 to 0.30) [94]. In a meta-analysis of 9 studies by Dineva et al., no differences were found in the incidence of hyponatremia in patients receiving hydrochlorothiazide or chlorthalidone [95].
Based on the results of clinical studies, factors predisposing to TIH were identified. In a meta-analysis of 43 clinical trials completed by Barber et al. and including 3269 patients with TIH, it was shown that the factors predisposing to TIH were: old age and low body weight of patients [96]. On the other hand, in the study by Huang et al., which included 48 patients with TIH and 211 patients with normal natremia treated with thiazide or thiazide-like diuretics, the clinical and genetic features were characterized. In a multivariate analysis, it was found that increased risk of hyponatremia in the elderly (aged 77.5 ± 9.2 years) (OR = 1.13; 95% CI: 1.08–1.19, p < 0.001) and women (OR = 4.49; 95% CI: 1.54–13.11, p = 0.008) was found. It was also shown that some polymorphisms of the KCNJ1 gene (potassium inwardly rectifying channel subfamily J member 1) and ROMK (renal outer medullary potassium channel) gene highly expressed in epithelial cells of the thick-walled part of the ascending loop of Henle and in the collecting duct) significantly increased the risk of TIH (OR = 5.75; 95% CI: 1.25–26.45, p = 0.03) [97]. A study by Ware et al. showed a significant relationship between a single nucleotide polymorphism in the SLCO2A1 (solute carrier organic anion transporter family member 2A1) (A396T) gene encoding the prostaglandin transporter (PGT) and the risk of TIH (OR = 3.33; p = 0.0005) [98]. Other risk factors for TIH include hypokalemia and the onset of use of thiazide or thiazide-like diuretics during the summer [96, 98–102].
TIH: clinical characteristics, diagnostic criteria and prevention
The previously cited meta-analysis by Barber et al. showed that in patients using thiazide or thiazide-like diuretics, TIH occurs most frequently, in 50-90% of patients, during the first 19 days after starting treatment (95% CI: 7.9–30 days) [96]. However, it should be noted that TIH might occur at any time during the use of thiazide or thiazide-like diuretics. A study by Friedman et al. has shown that in patients susceptible to TIH, natremia is reduced within a few hours after the administration of thiazide or a thiazide-like diuretic, and TIH may occur within the first 48 h of treatment [103]. It should also be emphasized that TIH in susceptible patients recurs after re-administration of thiazide or a thiazide-like diuretic. In addition, any thiazide or thiazide-like diuretic can induce TIH (TIH has been reported with hydrochlorothiazide, bendofluazide, indapamide and chlorthalidone use). TIH is more common with high doses of thiazide or a thiazide-like diuretic, but in approximately 10% of patients, TIH was caused by the administration of 12.5 mg hydrochlorothiazide only [96, 99–102].
Hyponatremia due to thiazide or thiazide-like diuretics is usually profound hyponatremia. In the meta-analysis by Barber et al., it was found that the mean serum sodium concentration in patients with TIH was 116.4 mmol/l (95% CI: 113.4–119.5 mmol/l). The decreased natremia was accompanied by hypokalemia ([K+] = 3.3; 95% CI: 3.0–3.5) [95]. The same meta-analysis found that the most common symptoms in patients with TIH were: neurological symptoms (51%), falls (48%), fatigue (46%), weakness (45%), confusion (44%) and nausea (36%) [100].
TIH diagnostic criteria include normal hydration status by physical examination (no evidence of overhydration or volume depletion), increase of decreased natremia after discontinuation of thiazide or a thiazide-like diuretic (by 3 mmol/l after 1 day or 5 mmol/l after 2 days), no increase of decreased natremia in the case of continued use of thiazide or a thiazide-like diuretic and no recurrence of hyponatremia after discontinued use of thiazide or a thiazide-like diuretic [99].
In order to prevent TIH, the symptoms of its occurrence should be monitored and the patient should be educated in this regard. Patients treated with thiazide or thiazide-like diuretics should be informed about the symptoms of TIH and about the need for an urgent medical consultation if they occur. Moreover, it is advisable to avoid excessive water intake (i.e. > 2.5 l/day) in patients treated with thiazide or thiazide-like diuretics. Thiazide and thiazide-like diuretics should not be used in patients with a history of TIH. In clinical situations with the need of a diuretic, a loop diuretic should be recommended. In patients using thiazide or thiazide-like diuretics, it is suggested to measure natremia at least 2-3 weeks after starting treatment, and then at least once a year [17, 99, 101, 102].
TIH: pathogenesis
The pathogenesis of TIH is not fully understood. TIH shows increased water reabsorption in the collecting tubules, decreased free water excretion, increased translocation to the AQP-2 cell membrane and increased body weight due to increased water content in the body. In addition, an increase in the urinary excretion of prostaglandin E2 (PGE2) is observed. About 50% of patients with TIH have decreased PGT activity in the collecting tubules caused by a mutation of the SLCO2A1 gene [23, 97, 99, 100, 103–105]. Under physiological conditions, PGE2 is secreted into the kidney’s interstitium and into the lumen of the collecting ducts due to an increase in volemia. From the lumen of the collecting ducts, PGE2 is transported back to the cytoplasm of cells with PGT and further to the renal interstitium, also by this transporter. Prostaglandin E2 then autocrine stimulates EP1 (prostaglandin E2 receptor 1) and EP3 (prostaglandin E2 receptor 3) receptors located on the cell membrane directed to the renal interstitium (abluminal) of collecting duct cells, leading to a reduction in the amount of AQP2 in the luminal membrane of these cells. The effect of PGE2 under physiological conditions is therefore an increase in diuresis and a decrease in volemia (Figure 5 A) [104]. In patients with the SLCO2A1 gene mutation, PGE2 is not efficiently transmitted by PGT after administration of thiazide or a thiazide-like diuretic, and probably, under the influence of these drugs, its production in collecting ducts is increased. In the lumen of the collecting ducts, PGE2 stimulates EP4 (prostaglandin E2 receptor 4) receptors, causing an increase in the production of cyclic adenosine monophosphate (cAMP) in these cells, which leads to increased AQP2 translocation to the luminal membrane of collecting ducts, which results in increased water resorption (Figure 5 B) [104].
Figure 5
A – The physiological role of PGE2 in the regulation of volemia; B – Pathogenesis of hyponatremia induced by thiazide or thiazide-like diuretics, caused by mutation of the prostaglandin transporter gene and their increased production in the nephron collecting duct cell
PGE2 – prostaglandin E2, PGT – prostaglandin transporter, EP1 – prostaglandin E2 receptor 1, EP3 – prostaglandin E2 receptor 3, EP4 – prostaglandin E2 receptor 4, AQP2 – aquaporin 2.
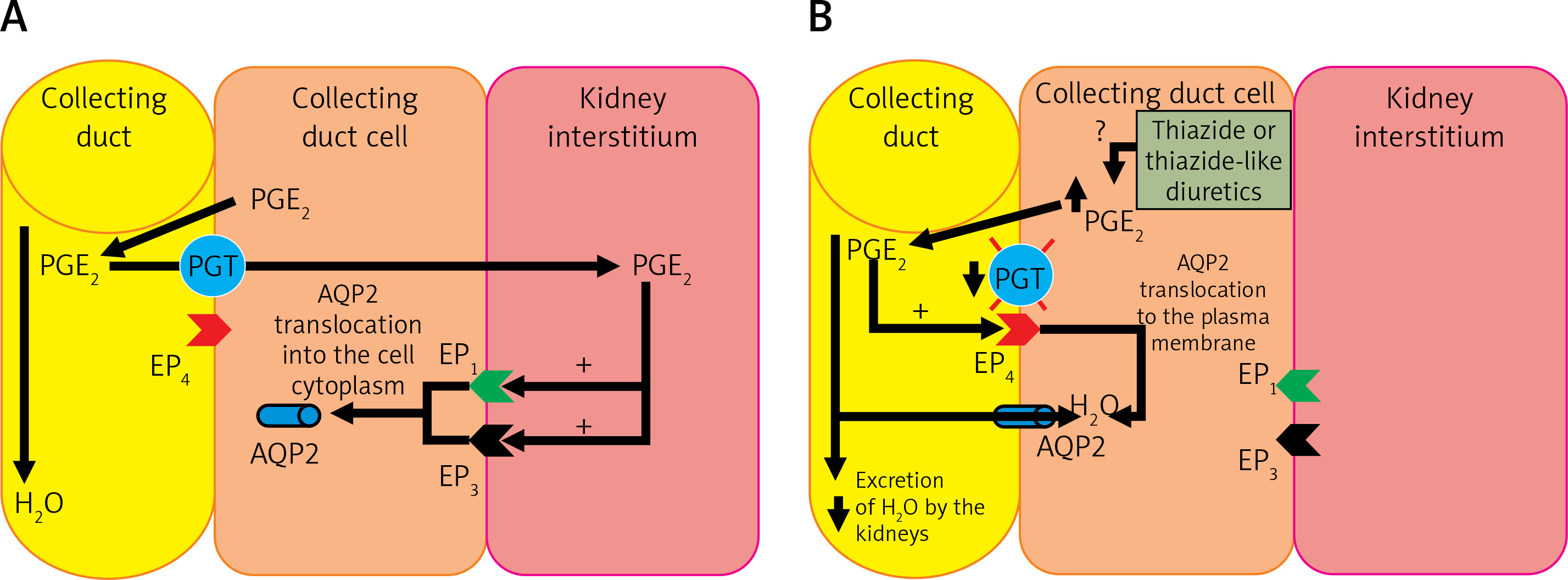
Thus, a significant proportion of patients with TIH experience excessive water reabsorption, leading to profound hyponatremia accompanied by hypokalemia [104]. It is worth noting that cells that clinically express the mutation of the PGT transporter gene (in the collecting duct) are located in a different part of the nephron than the target point of thiazide and thiazide-like diuretics (distal tubuli) [103].
Treatment of hyponatremia
In the treatment of hyponatremia, the guidelines published (for example, the European Renal Best Practice, ERBP) should be followed [16].
In the case of acute hyponatremia with symptoms of injury to the CNS (i.e. disturbances in perception and motor coordination, nausea and vomiting, confusion, headache, abnormal sleepiness, convulsions, and coma), immediate intravenous administration of 150 ml of a 3% NaCl hypertonic solution within 20 min followed by determination of plasma sodium concentration is recommended. The goal of treatment is to increase the natremia by 5 mmol/l. Subsequent intravenous infusions of 150 ml of 3% NaCl solution should be administered until the concentration of sodium in plasma is increased by 5 mmol/l [16].
In the case of an increase in natremia by 5 mmol/l in the first hour and clinical improvement of the patient’s condition, it is recommended to discontinue the administration of intravenous hypertonic NaCl solution. The increase in natremia during the first 24 h of treatment must not exceed 10 mmol/l, and in the following days, 8 mmol/l/day, until the target concentration of 130 mmol/l is achieved. The concentration of sodium in plasma should be determined after 6 and 12 h, and then once a day during treatment until a stable concentration of sodium in the plasma is obtained [16].
In the event of a 5 mmol/l increase in natremia in the first hour and no clinical improvement in the patient, it is recommended to continue intravenous administration of a hypertonic 3% NaCl solution to obtain an additional 1 mmol/l/h increase in natremia. In the case of improvement of clinical status or a total increase in natremia by 10 mmol/l, and also when the concentration of sodium in the plasma reaches 130 mmol/l, the intravenous administration of a hypertonic 3% NaCl solution should be discontinued. If the symptoms of injury to the CNS persist, it is necessary to extend the diagnostic procedures to look for other possible causes of the above-mentioned clinical symptoms, in addition to hyponatremia. Plasma sodium concentration should be determined every 4 h during the intravenous administration of 3% NaCl solution [16].
In patients with acute hyponatremia with symptoms of CNS injury, it is necessary to withhold, at least temporarily, all medications that may contribute to or worsen hyponatremia [16].
In the case of chronic hyponatremia, fluid and drug administration, which might lead to hyponatremia, should be discontinued, and the action of other factors that may aggravate the hyponatremia should be eliminated. Causal treatment of hyponatremia is recommended. In mild hyponatremia, aggressive treatment solely aimed at increasing the plasma sodium concentration (i.e. intravenous administration of 3% NaCl solution) is not recommended. In moderate to profound hyponatremia, it is recommended to avoid an increase in plasma sodium > 10 mmol/l on the first day and > 8 mmol/l on the following days. In moderate or profound hyponatremia, it is recommended to measure sodium concentration in plasma every 6 h until it reaches 130 mmol/l [16].
In patients with SIADH syndrome, fluid restriction is suggested as the first-line treatment. If such a procedure is ineffective, urea oral preparations should be used [16]. The daily dose of urea should be between 0.25 and 0.50 g/kg bw. To reduce the bitter taste of urea, add sweeteners. You can use a powder with the following composition: 10 g urea + 2 g sodium bicarbonate + 1.5 g citric acid + 200 mg sucrose. The contents should be dissolved in 50–100 ml of water before consumption [16].
A too rapid increase in plasma osmolality (mainly determined by an increase in natremia) leads to the displacement of water from the intracellular space to the plasma and to a decrease in the volume of cells (including cells in the CNS) and, consequently, to the osmotic demyelination syndrome (Figure 6).
Figure 6
Pathogenesis of cerebral edema in the course of hyponatremia and osmotic demyelination syndrome in the course of too rapid correction of hyponatremia
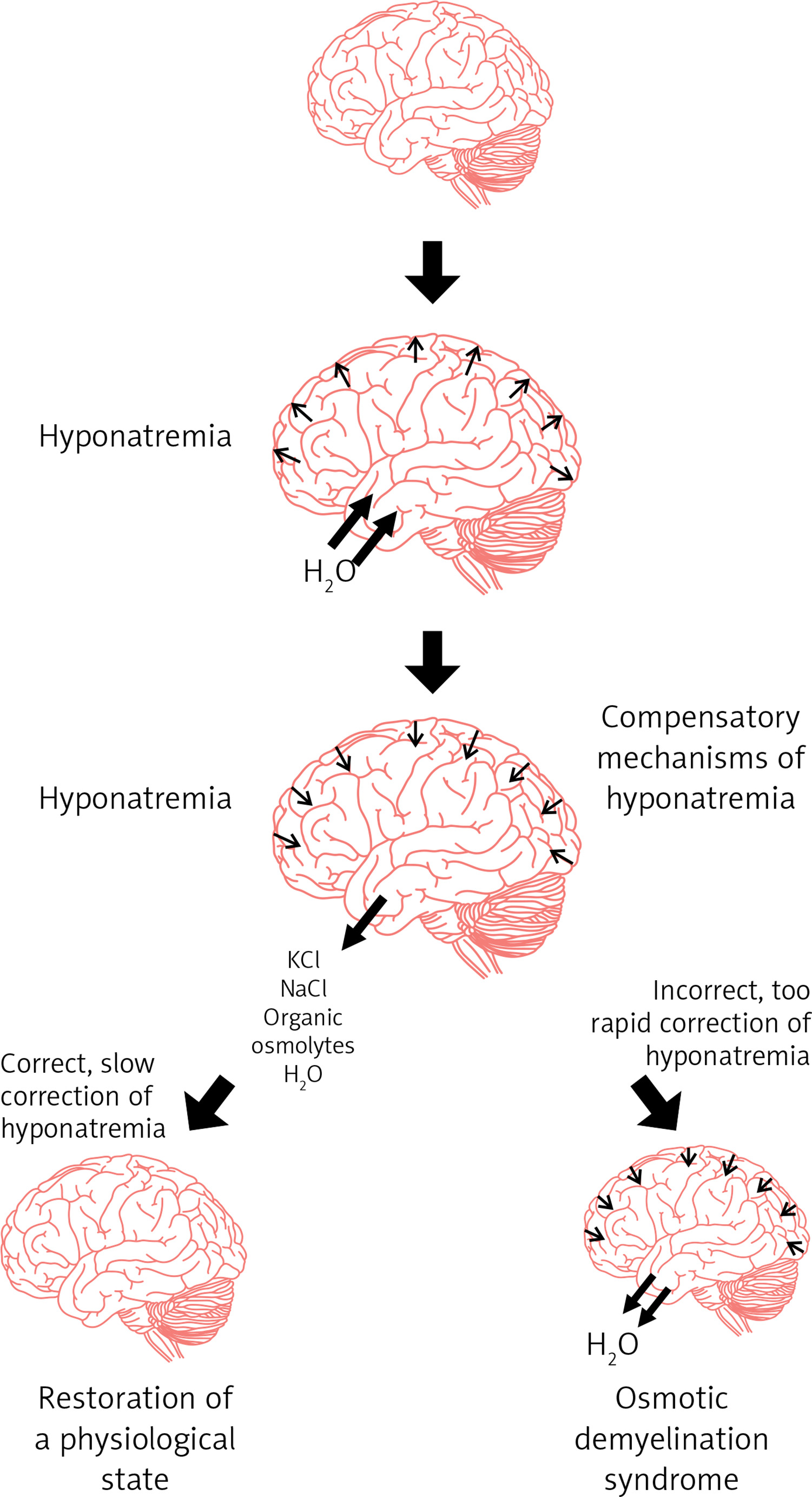
The symptoms of this syndrome are: dysphagia, dysarthria, behavioral disturbances, convulsions, tetraplegia, coma (Figure 7) [106, 107]. The risk factors that predispose the patient to the development of osmotic demyelination syndrome include: liver failure, alcoholism, malnutrition and hypokalemia. Osmotic demyelination syndrome usually leads to death [1, 15].
Figure 7
Correct (slow) and incorrect (too fast) correction of natremia during hyponatremia treatment and the occurrence of neurological complications in subjects with hyponatremia. Based on [106]
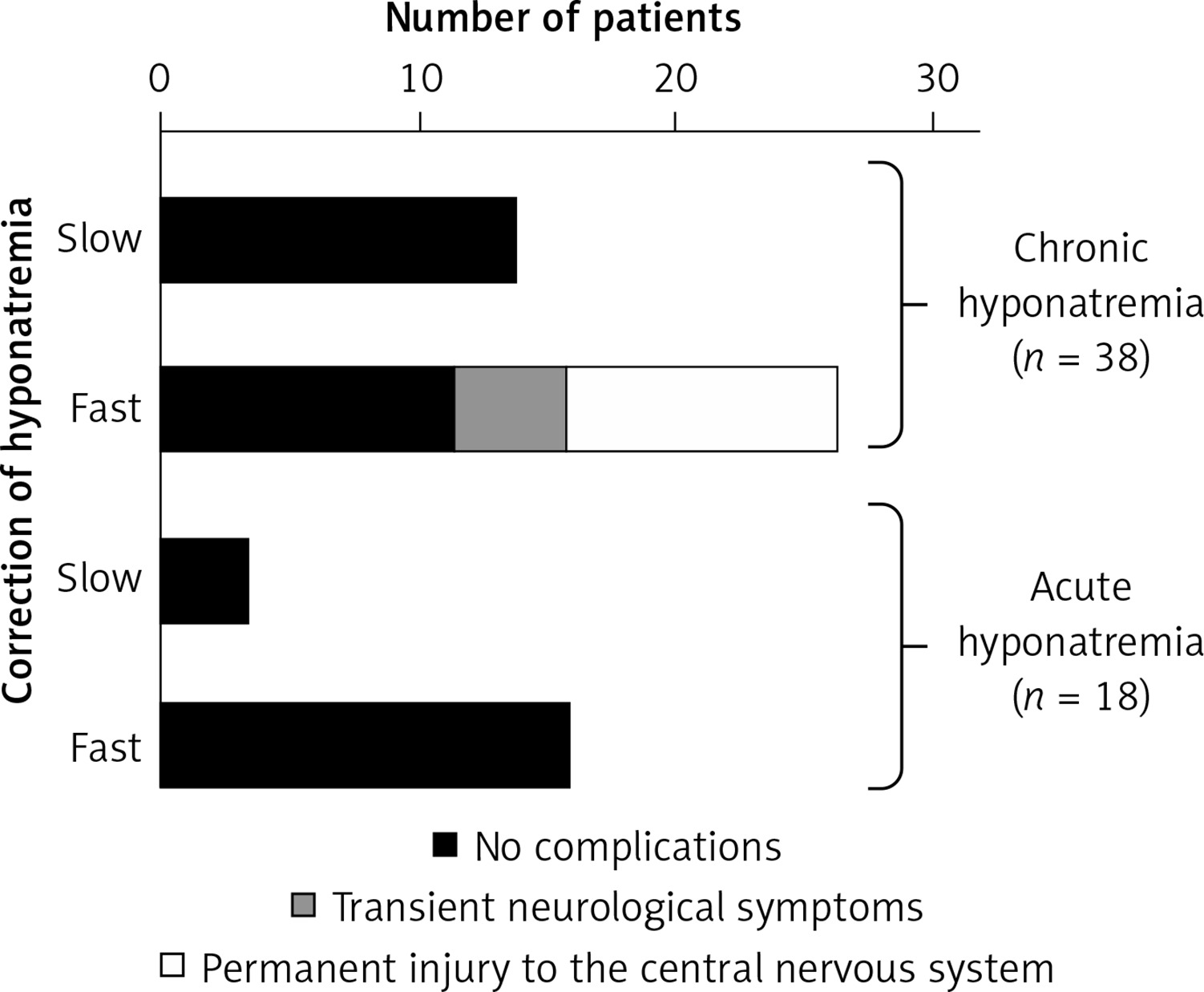
In case of too rapid correction of natremia, intervention to re-reduce plasma sodium concentration should be recommended. If natremia increases > 10 mmol/l on the first day or > 8 mmol/l/day on the following initiation of intravenous infusion of electrolyte-free fluid (e.g. water 5% glucose solution) at a dose of 10 ml/kg bw should be considered for 1 h [16].
Conclusions
Hyponatremia is defined by a plasma sodium concentration < 135 mmol/l and may be associated with features of volume depletion, normal hydration or hyperhydration. Hyponatremia is a common electrolyte abnormality in clinical practice and its occurrence adversely affects the prognosis of patients. Acute symptomatic hyponatremia within 48 h is life-threatening and requires immediate treatment. The most common cause of hyponatremia is SIADH. In patients with arterial hypertension, hyponatremia may be caused by the use of thiazide or thiazide-like diuretics. Too intensive treatment of hyponatremia may lead to neurological disorders associated with irreversible injury to the central nervous system in the form of the osmotic demyelination syndrome.