Introduction
The Arab population or people speaking the Arabic language are reported to have a significantly high incidence of genetic abnormalities due to the high rate of first-cousin marriages [1]. Increased level of inbreeding, large family sizes, and high mother and father ages characterize the Arab population. In earlier days not much familiar public health effort was aimed at preventing genetic diseases, but recently country-wide initiatives for screening and preventive measures have been initiated. In the recent past, many genetic disorders have received attention in the field of medical genetics to identify disease-causing mutations using next-generation sequencing techniques in the Arab region. Vast deserts, suffocating heat, oil wells, throngs of pilgrims, and prosperous-looking cities continue to dominate popular opinions of the Arab population, especially Saudi Arabia. This view is now expanded to incorporate an advanced initiative that is claiming a global spotlight as one of the top human genomics projects: the Saudi Human Genome Program (SHGP), Kuwait Genome Project (KGP), Qatar Genome Programme (QGP), Emirati Genome Program (EGP), and Bahrain Genome Project (BGP). Arab countries such as the Kingdom of Saudi Arabia, United Arab Emirates and Qatar have invested a lot of money and efforts in next-generation genomics to reveal the molecular basis of diseases and disorders prevalent in the Arab population. The post-Arab genome programs are a golden era in molecular genetics and disease biomarkers of Arabian genomic studies. These Arab genome programs have overcome the obstacles and limitations of collecting samples. The SHGP, KGP, QGP, EGP and BGP programs are visionary projects on human genomics with the vision to detect and prevent genetic diseases in the Kingdom of Saudi Arabia (KSA), the State of Kuwait, the State of Qatar, the United Arab Emirates (UAE) and the Kingdom of Bahrain, to establish world-class facilities in genomic research in the Arab ancestry [2–6]. The SHGP was implemented by King Abdulaziz City for Science and Technology, on its way to sequence 100,000 human genomes in Saudi Land to achieve gold standard findings in the Saudi population on genomics-based biomedical research [2]. The central laboratory of SHGP in 2018 is one of the first national initiatives and the largest genomic project in the Middle East; details can be obtained from the freely available database (https://shgp.kacst.edu.sa/index.en.html) [7]. The SHGP initiative revealed > 3000 new nucleotide variants to the literature, that are associated with > 1200 rare genetic disorders [7–9]. Earlier reviews on population structure and prevalent genetic disorders can help to understand the initial studies on Mendelian genes and associated traditions in the high consanguineous community of the Kingdom of Saudi Arabia and other Arab populations [10–12]. The State of Qatar, which is located in the Arabian Gulf, established the QGP (https://www.qatargenome.org.qa/), a Qatari population-based genome initiative aimed at sequencing the complete genomes of a large part of the Qatari population, which began its pilot phase in 2015 [3]. The Kuwait Genome Project (KGP) is a high-coverage effort to sequence Kuwaiti genomes from genetically different subgroups of Kuwaitis [4].
Although genome research has the potential to transform medical approaches related to prevention of disorders, developing advanced diagnosis, and formulating treatment strategies, it is not without its obstacles and limitations [3]. The willingness of individuals to volunteer samples and use genetic testing services is critical to achieve the goals of the national genome initiatives like KGP, SHGP, QGP, BGP and EGP. Basic genetic knowledge, a genetic history of families with genetic disorders, and prior experience were all found to be significant predictors of willingness of Arabs to participate. The findings from 837 adult Qataris show widespread acceptance, but the study also points to the need for greater education and personalized counselling to explain the procedure, obstacles, and advantages of genetic testing [3]. These Arab genome initiatives have surmounted the challenges and constraints of sample collection. Whole exome sequencing (WES) on evaluating 48 Qatari (30 families) patients with inherited Mendelian disorders from the “Qatar Mendelian Disease pilot program”, revealed 25 unique disease-causing pathogenic alleles that are important to premarital and newborn screening of Arab populations [13]. Next-generation sequencing methods such as WES play an important role in molecular pathogenesis [14, 15]. The narrative review reviews the advances throughout the Arab genome program period, including major breakthroughs in pathogenic disease-associated variations and advances in molecular genetics from large-scale whole-exome sequencing data.
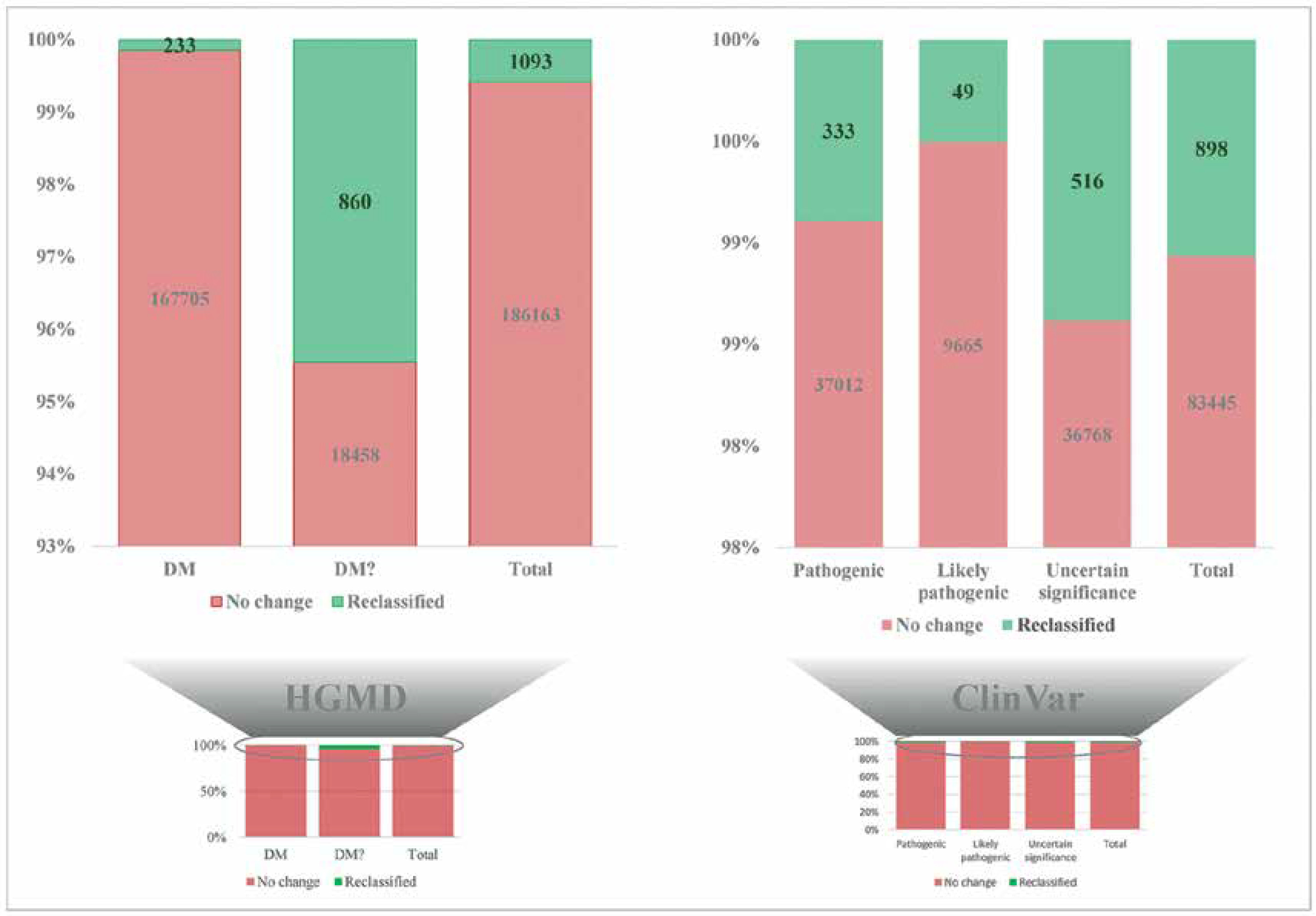
Novel variants and reclassifying pathogenic variants as benign
The initial study on Qataris revealed 58.37% novel variants among the identified 5,452,613 variants [16]. Significant differences were observed in the initial studies on the Saudi genome [17], and Kuwaiti genome [4, 18]. Still more in-depth genome-wide studies are needed [19]. Recent findings from QGP phase 1, which sequenced the whole genomes of 6,045 Qataris, were reported. They found almost 88 million DNA variations, interestingly, with 24,620,313 of them being novel to various genome databases [20]. The study discovered that many uncommon deletion variations were more common and prevalent in the Qatari population. Most of the observed novel variants appear to give disease protection in the Qatari population and have changed the genetic architecture, which is consistent with the region’s high consanguinity. Five non-admixed groupings were discovered in the genetic makeup of the Qatari population. The study also described the heritability of genetic disease marker correlations for 45 clinical phenotypes [20]. A recent study on reclassification of pathogenic variants (16 Human Gene Mutation Database (1 disease mutation and 15 disease mutations) variants and 10 ClinVar (6 pathogenic, 4 variants of uncertain significance) variants) as benign by the team of the Saudi Human Genome Program (SHGP) database was possible because of the world’s biggest collection of genetic variations from Arabs through the establishment of the SHGP database [8]. The SHGP database using 3070 disease-associated genes (including 2000 whole-exome sequencing WES) covering > 4000 Mendelian disorders of 5849 non-overlapping individuals can reveal Arab-enriched and Arab-specific common variants in the Arab population in general and the Saudi population in particular [8]. It was expected that at least some reported “genetic disease-associated variants” might characterize Arab-enriched or Arab-specific common mutations that might not be detected as such by utilizing these genomic data as most of the disease-associated genes and variants were described for the first time in Arab subjects with disease phenotype due to the high level of consanguinity. Also these factors enable positional mapping in the genome, since ethnicity is not much represented in publicly accessible variant databases (Figure 1). The study revealed that 16 HGMD variants and 10 ClinVar variants had a Saudi ancestry frequency (in population) of 5% in the database (SHGP) but only < 5% in publicly available databases, allowing all these 26 pathogenic variants to be reliably reclassified as benign. Autozygosity is rich in the SHGP database; based on Saudi population frequency (> 1%) and occurrence of genotype homozygosity in Saudi individuals who lack the phenotype described already, the study was able to reclassify 484 ClinVar variants (184 pathogenic, 25 likely pathogenic, 275 variant of uncertain significance) and 607 HGMD variants (103 disease mutation and 504 disease mutation) as benign [8]. Due to the founder effect, QGP revealed certain rare pathogenic variant mutations with high MAF in the Qatari population [20]. Variant rs750046020 in the MPL gene, specifically linked with thrombocytosis, occurs with MAF of 0.009 in the Qatari Arab population [20]. Similarly, at an MAF of 0.007, variations in the genes KRT5 (rs267607448) and CBS (rs398123151) linked to epidermolysis bullosa and homocystinuria, respectively, are found in the Qatari population [20]. Ten novel mutations were discovered in ten known genes associated with myeloproliferative neoplasms, and seven potential candidate genes were discovered with seven novel variants from Qatari patients with myeloproliferative neoplasms [21]. A total of 1,790,171 annotated variants and 8,462 structural variants, found as novel variations across the Emirati genome, were found while constructing the United Arab Emirates Reference Genome [22]. Kuwaiti genome studies discovered 58,186 SNPs, 32,686 indels as novel variants [4, 18]. Most of the coding novel variants discovered in the Kuwaiti genome studies were found to be associated with autosomal recessive disorders in the Arab population [18].
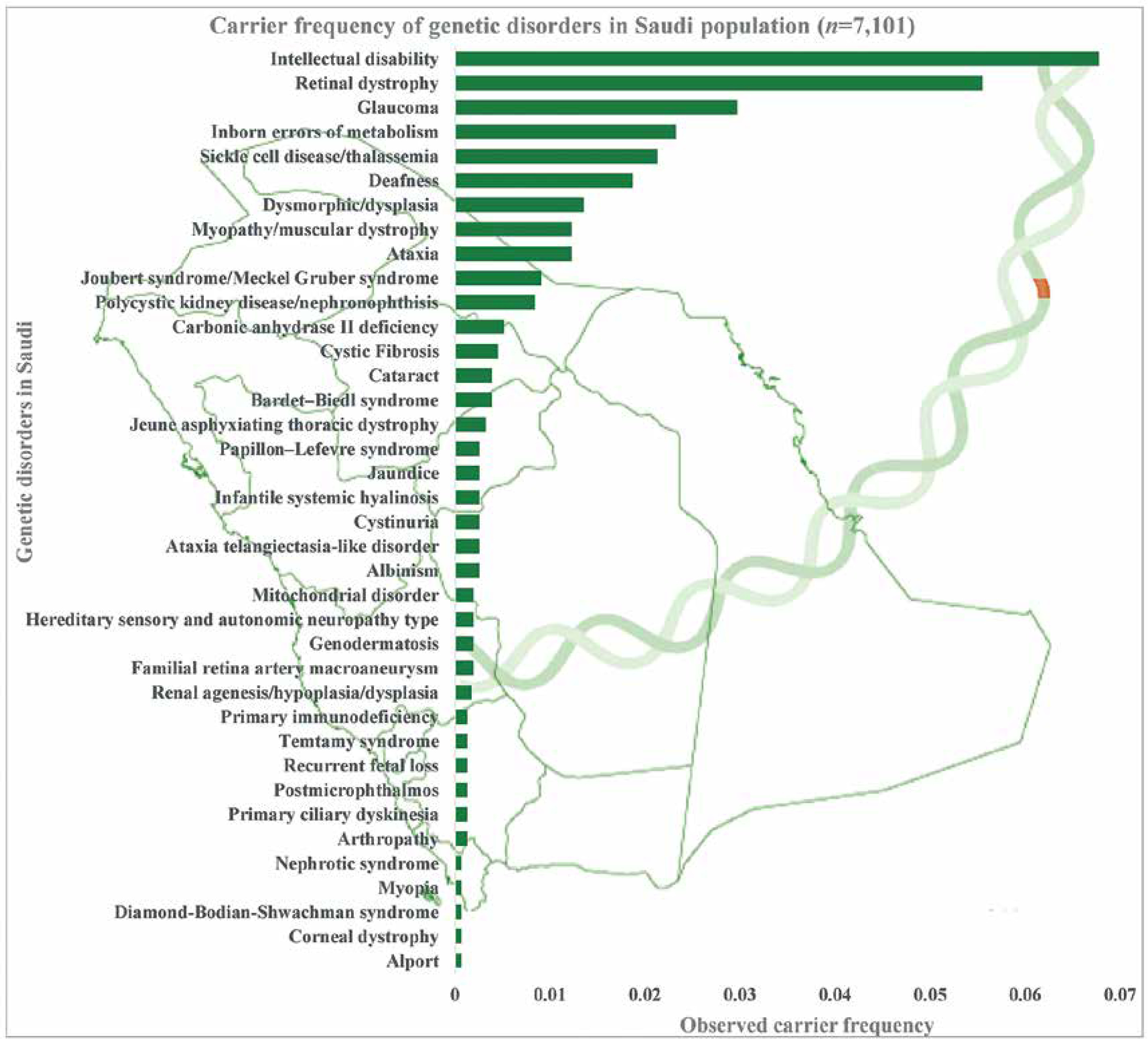
Carrier frequency of genetic disorders
The combined and observed carrier frequency of genetic disorders among Saudi Arabians was estimated for the first time by utilizing a large sample (n = 7,101 patients) using targeted gene panel sequencing and whole-exome sequencing [7]. The study revealed the most prevalent carrier status of various diseases (Figure 2): intellectual disability comes first with the combined and observed carrier frequency of 0.06779, followed by retinal dystrophy, glaucoma, inborn errors of metabolism, sickle cell disease/thalassemia, deafness, dysmorphic/dysplasia, ataxia, myopathy/muscular dystrophy, polycystic kidney disease/nephronophthisis, Joubert syndrome/Meckel-Gruber syndrome, carbonic anhydrase II deficiency, cystic fibrosis, Bardet-Biedl syndrome, and cataract [7] (Figure 2). Furthermore, a study on 2357 Saudi patients through gene panels revealed 433 novel disease alleles and 355 existing variants, which is the largest notable number of novel disease alleles submitted to the HGMD in a single genomic study. Third, the 433 novel disease variants identified in the genome study by the Saudi Mendeliome Group represent an exceptional resource on the Arab variome (whole set of genetic variations) as most of the study participants were of Arab origin. Hence these observations and additions to the HGMD by the Saudi genome studies constitute an invaluable resource for the interpretation of clinical and molecular genomic analysis on Mendelian genes in Arab patients [23]. These novel disease markers will help address the uncertainty in the identification of many Arab-specific disease markers. All these studies reveal a high prevalence of variants and carriers, as the degree of consanguinity is high among the Saudi ancestry; hence studies were able to identify a lot of autozygosity-related variations in homozygosity [7, 23]. The large-scale exome studies on the carrier frequency of genetic disorders among the Arab population change the previous observations of the most prevalent hemoglobinopathies [7, 11]. Carrier frequency of the intellectual disability is three times more than that of sickle cell disease and thalassemia among the Arab population with 25–60% consanguinity rates [7, 11]. In-depth functional analysis of observed intellectual disability genetic markers can help in the early identification of intellectual disability genetic markers and designing novel treatment strategies. The most prevalent recessive alleles discovered in the Qatari population are those associated with developmental disorders and structural deformities [20]. It is not surprising that the large number of Arabs in Saudi Arabia share similar and common carrier frequency with Qatar’s native population with minor differences in some alleles (DCAF17: c.436delC) [20].
Clinically relevant traits and precision medicine
A foundation stone was laid in studies on the Qatari population on the clinically relevant traits and loci associated genetics along with earlier studies [16, 24]. Genetics has an important role in the output and diversity in the clinical laboratory test results and decision-making in precision medicine [24]. Furthermore, very little is known about the genetic variability of the Arab population compared to other ancestries. A detailed study presented the results of genome-wide association research on forty-five clinically relevant features among the Qatari population, conducted by means of WES technique of 6218 subjects [24, 25]. They reported Arab (Qatari) and European populations with similar trait heritability (r = 0.81). Moreover, Arab (Qatari) ancestries are less similar to African ancestries (r = 0.44) in trait heritability. The authors found 281 unique and significant associations between the variants and clinical traits through GWAS analysis, which coincide with previously discovered associations with other populations, mainly Europeans. The relationships between allele frequencies for repeated loci are lower in Japanese (r = 0.80) and African (r = 0.85) populations while being higher in European (r = 0.94) populations [26–29]. The authors presented 17 novel and Arab-dominant signals based on Qataris that shed light on the molecular pathways that control the clinical traits [24]. The polygenic scores developed from European data have lower predictive performance in the Qatar-based Arab population. Findings from the large-scale analysis on the clinical traits of the Arab population are expected to have an impact on its future use in precision medicine and genetic architecture of complex diseases prevalent in the Arab population. Finally, for the first time in an Arab population, this study on heritability analysis and GWAS-based genetic association analysis on clinically relevant variables importantly identified shared genetic loci with variation in linkage disequilibrium patterns [24]. Metabolic quantitative trait loci identification analysis revealed genetic loci associated with pharmacological/drug targets on the consanguineous Arab population, which is important for precision medicine in the Arab world [30]. Constructing specific reference sequences for a subpopulation of Arab ancestry is also important in precision medicine; the initiation of the UAE reference panel is a good initiative [22, 31, 32].
High genetic heterogeneity
The Arab population is known for its high genetic heterogeneity in the diversity of phenotypes and clinical outcomes [1, 33–38]. The Arab world is an ethnocultural melting pot located geographically as a land bridging Eurasia and Africa, which functioned as a gateway for the migration of humans from the African region to the rest of the civilized world. The wide range of pathogenic gene mutations linked to various genetic diseases in the Arabs perhaps best exemplifies the heterogeneity of Arabs [1]. The high consanguinity of Arab people is well known. Because of the complexity of consanguinity and heterogeneity, understanding the causes of Arab population-specific genetic disorders in the region is challenging. The Arab region is populated by genetically diverse populations, as evidenced by previous genetic studies [1, 33–36]. Inter-regional genetic variability in Arab ancestry is also highlighted in a recent GWAS study [36]. The prevalence of heterogeneity in genetics of highly consanguineous Arab families makes the interpretation for the genetic basis and etiology of numerous inherited disorders most common in Arab populations. In Arab countries, more than 1200 distinct genetic disorders have been documented, 60% of which are autosomal recessive disorders and more than 40% are confined to a certain demographic or geographical location [1, 10–12, 33]. High-resolution population genetic studies on the Arab population were carried out to understand the genetic diversity of inherited diseases and the degree of heterogeneity among Kuwaiti Arabs using SNP microarray (n = 583) [1]. Within the newly established groupings, the study infers the primary sources of ancestry among Kuwaiti Arabs. Various findings reinforce the genetic imprints and regional genetic heterogeneity in Arab people on the blood disorders, metabolic disorders, disorders of the circulatory system, breast cancer, ovarian cancer, colorectal carcinoma and prostate cancer [1].
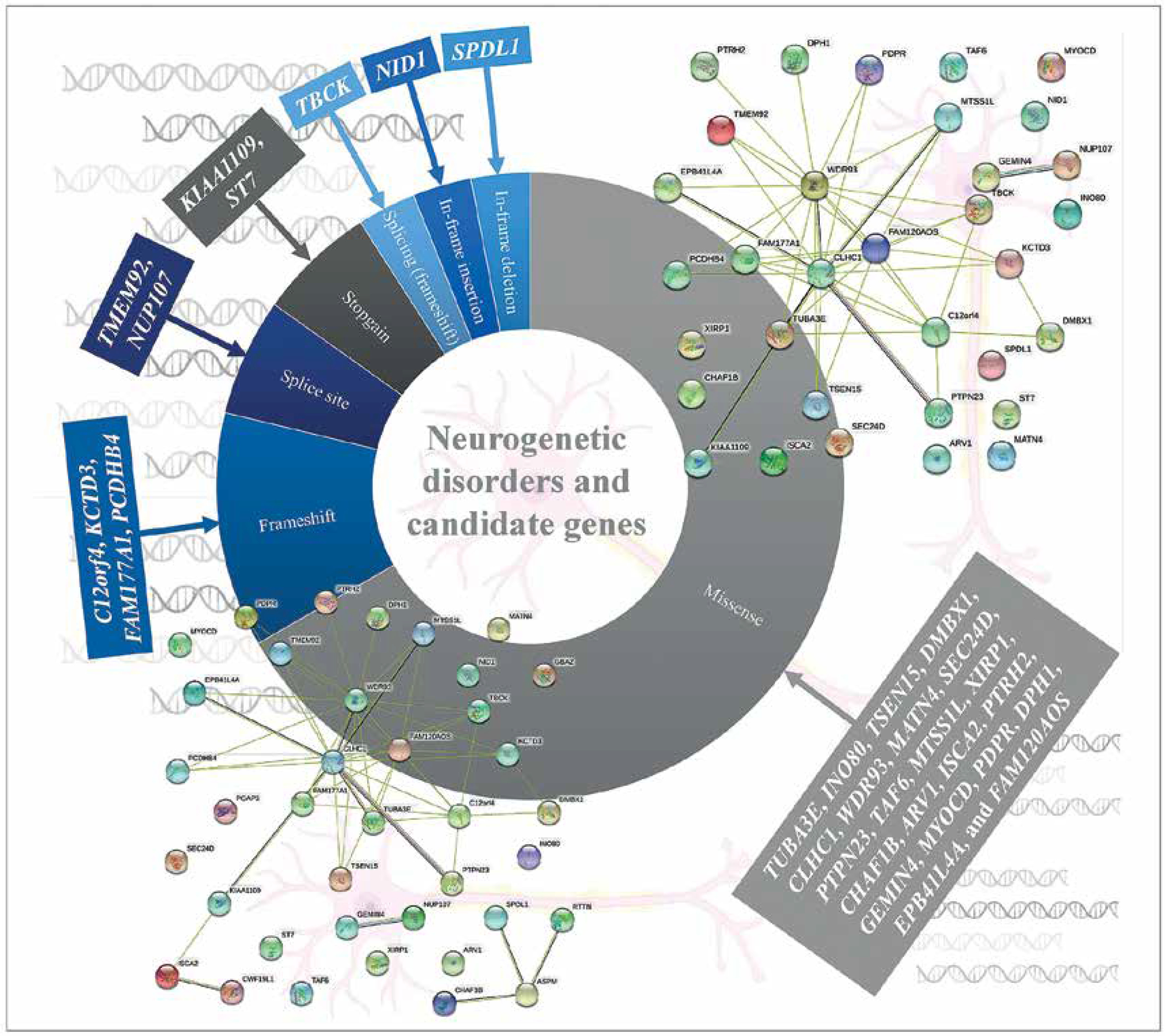
Neurogenetic disorders and candidate genes
The most common referral request for WES analysis was for neurological diseases in an Arab country (Lebanon) [39]. WES analysis of pre-screened multiplex consanguineous families with neuro-genetic disorders revealed candidate genes and disease markers [40]. 33 genes (observed phenotype), i.e. SPDL1 (primary microcephaly and neonatal death), TUBA3E (microlissencephaly and global developmental delay), INO80, TSEN15, and PCDHB4 (global developmental delay or GDD and primary microcephaly), DMBX1 (epilepsy, GDD, and poor weight gain), CLHC1 (myopathy), NID1 (muscle weakness, hydrocephalus, and global developmental delay), C12orf4 (global developmental delay), WDR93 (autism spectrum disorder), ST7 (brain atrophy and global developmental delay), MATN4 (holoprosencephaly), SEC24D, and MYOCD (intellectual disability and epilepsy), KIAA1109 (Dandy-Walker malformation, club feet, flexed deformity, hydrocephalus, pleural effusion and micrognathia), PTPN23 (global developmental delay and brain atrophy), TAF6 (global developmental delay and dysmorphism), TBCK (hypotonia, GDD, dysmorphism, epilepsy, and VSD), FAM177A1 (macrocephaly, dolichocephaly, intellectual disability, and mild obesity), MTSS1L (neurodegeneration and brain iron accumulation), ARV1 (neurodegenerative disease), XIRP1 (primary microcephaly), CHAF1B (global developmental delay and ADHD), KCTD3 (cerebellar hypoplasia, seizure, and severe psychomotor retardation), ISCA2 (consistent with mitochondrial encephalopathy and neurodegeneration with high lactate peak in the brain and marked white matter changes), PDPR (typical Joubert syndrome and GDD), PTRH2 (hearing loss, and ataxia also GDD), GEMIN4 (severe osteopenia congenital cataract, tubulopathy, and GDD), DPH1 (Dandy-Walker malformation, developmental delay, cerebellar vermis hypoplasia, and hydrocephalus), NUP107 (early onset focal segmental glomerulosclerosis, light complexion, and global developmental delay), TMEM92 (cerebellar atrophy, hydrocephalus; cognitive, speech, and motor delay), EPB41L4A (spastic paraplegia, failure to thrive), and FAM120AOS (GERD, chronic lung disease, coarse facial features, skin laxity, scoliosis, hypotonia, undescended testicles and pectus excavatum), were identified among the pre-screened multiplex consanguineous families with neurogenetic disorders. Missense (n = 22) variations were common among the disease variations in the patients with neurogenetic disorders (Figure 3) [40]. RTTN and ASPM genes were associated with primary microcephaly apart from those listed above [40–42]. Cerebellar ataxia in a sibling WES study showed a novel variant in the CWF19L1 gene [43] and GBA2 gene in a consanguineous Saudi family [44]. A study on Qatari subjects revealed the association of a novel pathogenic PGAP3 variant with global developmental delay and neuromuscular abnormalities and brain anomalies [45]. Studies on Egyptian subjects revealed the association of pathogenic SCN10A variation in patients with neuromuscular disease and epileptic encephalopathy [46], SLC39A8 variation in patients with intellectual disability, developmental delay, cerebellar atrophy, hypotonia, strabismus, and variable short stature [47], PQBP1 variation in patients with intellectual disability [48] biotin-thiamine responsive encephalopathy [49] and TTC5 variation in patients with intellectual disability syndromes [50]. The PUS3 gene with a frameshift variant was observed as a candidate gene for intellectual disability in a study with 103 families from Jordan [51]. A family WES study revealed variations in the genes ASPA and ARSA in patients with leukodystrophy in Jordan [52]. Trio exome sequencing of four families from Jordan identified a multi-exon deletion in the VPS13B gene in patients with Cohen syndrome [53]. Consanguineous families from Jordan with neuromuscular disorders identified multiple variants WES in DYSF, MPV17, SLC25A46 and SGCG genes [54]. Lebanese subjects with neurodevelopmental and neuromuscular disorders from 200 WES analyses and other studies revealed various genes with pathogenic variations [55–57]. In order to identify the associated pathways and biological processes associated with the genes identified as the neurogenetic disorders candidate genes of the Arab population, protein-protein interaction analysis was carried out using STRING 10 [58]. The analysis revealed that the genes are linked in terms of text mining, and no significant associated pathways were identified, which indicates that the impact of cellular processes and pathways needs to be studied to reveal the neurogenetic disorders’ candidate genes. Incorporation of proteomics and pathway analysis may reveal the impact of individual genes on neurogenetic disorders in the Arab population and the network of gene-associated pathways on neurogenetic disorders.
Blood and bleeding disorders
Molecular defects, blood disorders, β-thalassemia, sickle cell disorder, α-thalassemia and G6PD (glucose-6-phosphate dehydrogenase) deficiency are the most common in the Arab population; they were extensively listed and reviewed earlier [1, 59–63]. The large-scale (~5000) WES analysis by the SHGP on 1285 cases with 17 blood and bleeding disorders revealed more novel variants (n = 140) than previously reported pathogenic variants (n = 98) (Table I) among the 821 variants [9, 64–67]. VWF, F8, F5, G6PD, F2, F7, F10, F13B, FGA, and HBA2 genes were observed with novel variants. The authors prioritized the list of genes and novel variants in their study, which will definitely be a source of genetic data and will aid in the development of molecular variants’ screening and to enhance the efficiency of the preventive programs for the blood and bleeding disorders prevalent in the kingdom [9]. These molecular data will also have a significant impact on developing molecular diagnostic tools for the clinically overlapping disorders with blood and bleeding disorders and to design treatment strategies. VWF, F10, HBB, F7, and F5 genes were observed with notable carrier frequency on the variants associated with Saudi ancestry with blood and bleeding diseases. VWF, G6PD and F8 genes are considered as important genes of interest in the prevailing blood and bleeding disorders [9]. The study very precisely ranks the variants based on novelty, pathogenicity and existence in the genomic databases. The study suggested prioritizing autosomal recessive variants in HBB, F7, F13A1, F11 and VWF genes to screen the carriers for the blood and bleeding disorders prevalent in the Saudi population [9].
Table I
Novel variants observed and their significance in the Saudi population
| Study description | Previously reported pathogenic variants | Novel variants | Prominent genes with novel variants | Novel variants and notable phenotype | Reference |
|---|---|---|---|---|---|
| Blood and bleeding disorder | 98 (stop loss = 1, synonymous = 2, splice and UTR = 11, truncating = 12, variants of unknown significance = 19, benign or likely benign = 27, re-classified pathogenic or likely pathogenic = 52, CADD score > 20 = 56, nonsynonymous = 72) | 140 (nonsynonymous = 117, CADD score > 20 = 71, pathogenic = 32, splice = 6, truncating = 4, frameshifts = 13, likely benign = 3, variants of unknown significance = 105) | VWF, F8, F5, G6PD, F2, F7, F10, F13B, FGA, HBA2 | 3 truncating variants observed in 2 cases of aplastic anemia, and 1 patients with excessive bleeding | [9] |
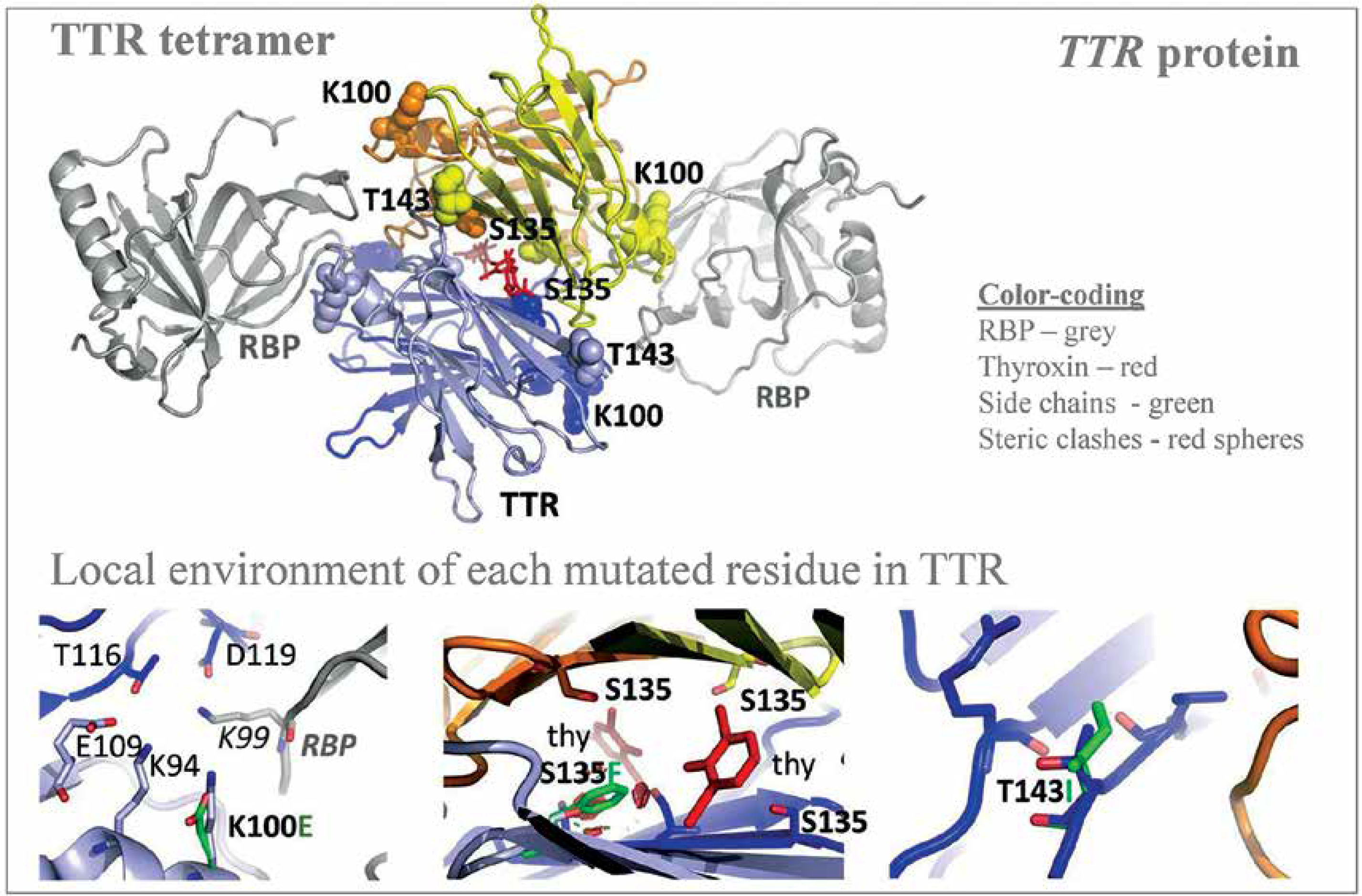
| Familial transthyretin (TTR) amyloidosis (ATTR) | 158 TTR variants (coding or flanking regions = 28, missense TTR variants = 12, uncertain significance = 3, negatively impact TTR function resulting in amyloidosis = 2) | 3 novel TTR c.404C>T (p.S135F), c.298A>G (p.K100E and c.428C>T (p.T143I), ) (nonsynonymous = 3, interface in the middle of the TTR tetramer and thyroxine = 1; interface in the middle of two TTR molecules and RBP4 = 1) | TTR | Only 1 allele of each novel TTR variant being observed | [64] |
| Antenatal cystic kidney disease and ciliopathy | 8 (CC2D2A, c.3084delG p.Arg1028Rfs*3; CC2D2A, c.3364C>T p.Pro1122Ser; CC2D2A, p.Trp1511Arg c.4531T>C; CEP290, c.5668G>T p.Gly1890*; PKHD1, c.4870C>T p.Arg1624Trp; RPGRIP1L, c.640G>A p.Val214Ile, c.685G>A p.A229T; TMEM231, c.751G>A p.V251I; CC2D2A, c.3084delG p.R1028Rfs*3) | 13 novel pathogenic variants (B9D1, c.508_510delCTC p.L170del; MKS1, c.417+1G>A; INVS, c.1760delA p.Q587Rfs*2; MKS1, c.1066C>T p.Q356*; TMEM67, c.457T>G p.C153G; TCTN2, c.1852C>T p.Q618*; TMEM67, c.1413-2A>G; MKS1, c.1066C>T p.Q356*; CEP290,c.3777_3778delAG p.R1259Sfs*16; PKHD1, c.3539G>A p.G1180E; NEK8, c.1401G>A p.W467*; CC2D2A, c.4437+1G>A; NPHP3, c.2694-1_-2delAG; TCTN2, c.252_253delTG) | B9D1, CC2D2A, INVS, MKS1, MKS1, TCTN2, TMEM67, CEP290, NEK8, NPHP3PKHD1, TCTN2 | Fetal death (B9D1, CC2D2A, INVS, MKS1); perinatal death (INVS, TCTN2); Ascites (CC2D2A, c.4437+1G>A); pericardial effusion and congenital heart malformation in Saudi ancestry (TMEM67, c.1413-2A>G); hepatomegaly (PKHD1, c.3539G>A) | [65] |
| Microphthalmia | 40 mutations | 15 novel | ALDH1A3, C12orf57, GDF3, CRYAA, DSC3, MAB21L2, OTX2, MFRP, MYO10, PAX6, PRSS56, PXDN, RAB3GAP2, SLC18A2, STRA6, ZNF219 | Variation in PRSS56 or MFRP cause posterior microphthalmos/nanophthalmos | [66] |
| Cholestatic liver disease | 22 mutations | 15 novel | ABCB11, ABCB4, ABCC2, AGL, ATP8B1, CYP7B1, FAH, TJP2, VIPAS39 | ABCC2, c.2273G> T with Dubin–Johnson syndrome; CYP7B1, c.1057G> T : p.E353X with bile acid synthesis defect | [67] |
Familial transthyretin amyloidosis
Autosomal dominant disease, familial transthyretin (TTR) amyloidosis (ATTR) was personalized for its genetic predisposition using 13906 Saudi exomes. The authors identified many amino acid substitution variants in the TTR gene and revealed the potential association with amyloidosis (Figure 4) [64]. From a total of 158 TTR mutations identified in the study population, 28 were in the coding and franchising region including 12 non-synonymous mutations during the analysis of 13906 Saudi exomes. Among the 12 missense TTR variants: three (c.239C>T:p.T80I; c.424G>A:p.V142I and c.238A>G:p.T80A) have a known impact on TTR functions; 3 (c.368G>A:p.R123H, c.385G>A:p.A129T and c.370C>T:p.R124C) have unknown significance; three (c.76G>A:p.G26S, c.140A>G:p.N47S and c.328C>A:p.H110N) mutations are benign and likely benign; and the remaining 3 (c.298A>G:p.K100E, c.404C>T:p.S135F, and c.428C>T:p.T143I) are novel variants [64]. Among the 12 missense TTR variants, 2 mutations [less frequent c.238A>G:p.T80A (0.00004), most prevalent c.424G>A:p.V142I (0.001)] are associated with amyloidosis, with a well-known pathogenic impact on the function of the TTR protein [64]. This study is the first large-scale study on analyzing the TTR mutations from the Saudi population. Even though three novel variants were reported in this study on Saudis with systemic amyloidosis, the known and previously reported mutation TTR:c.424G>A(p.V142I) is the variant observed most frequently in the Arabian ancestry [64]. This particular mutation is frequently identified in African descendants and the African American population. A common variant observed in Ireland and the United Kingdom, TTR:c.238A>G(p.T80A), is rare in the Arabian population [64]. Many variants of the TTR gene reported from Western Europe, Italian and Denmark populations were not observed in the Saudi ancestry; furthermore, the most common mutation observed in the TTR gene from Chinese and Mexican populations was also not observed in the Saudi ancestry including the 5 regions of Saudi Arabia. This study has a major limitation accessing the clinical data related to the novel TTR variants and the variance observed in the TTR gene, this very clearly indicates that more clinical and genetic studies are needed in order to reveal the clinical manifestations of the observed mutations in the Saudi ancestry.
Figure 4
Novel variants observed in TTR gene from Saudi population with familial transthyretin (TTR) amyloidosis (ATTR) or carrier. Modified from Abouelhoda et al. [64]. PDB ID: 2rox used for thyroxin-bound TTR and PDB ID: 2wqa used for RBP-bound TTR
Fetal death and perinatal death
A comprehensive genomic analysis on 34 families with fetal death, perinatal death and renal phenotype (enlarged, echogenic with cysts, cystic) revealed genes and novel variants associated with fetal death (B9D1, CC2D2A, and MKS1) and perinatal death (INVS and TCTN2) [65]. The study revealed 13 novel pathogenic variants [(Frame shift: INVS:c.1760delA; and CEP290:c.3777_3778delAG); deletions: B9D1:c.508_510delCTC; NPHP3:c.2694-1_-2delAG; and TCTN2:c.252_253delTG; CC2D2A: c.4437+1G>A; MKS1:c.417+1G>A; MKS1:c.1066C>T; TCTN2:c.1852C>T; NEK8:c.1401G>A; TMEM67:c.457T>G; TMEM67:c.1413-2A>G; MKS1:c.1066C>T; PKHD1:c.3539G>A); this clearly indicates the genetic heterogeneity. In additions to renal defects, ascites (CC2D2A), pericardial effusion and congenital heart malformation (TMEM67), and hepatomegaly (PKHD1) were also reported [65]. The most prevalent cause of an antenatal ciliopathy is due to CC2D2A gene variation among the families studied. Variations in FRAS1 and FREM2 genes were identified in six consanguineous Saudi Arabian families with antenatal/perinatal death [66–68].
Intellectual disability
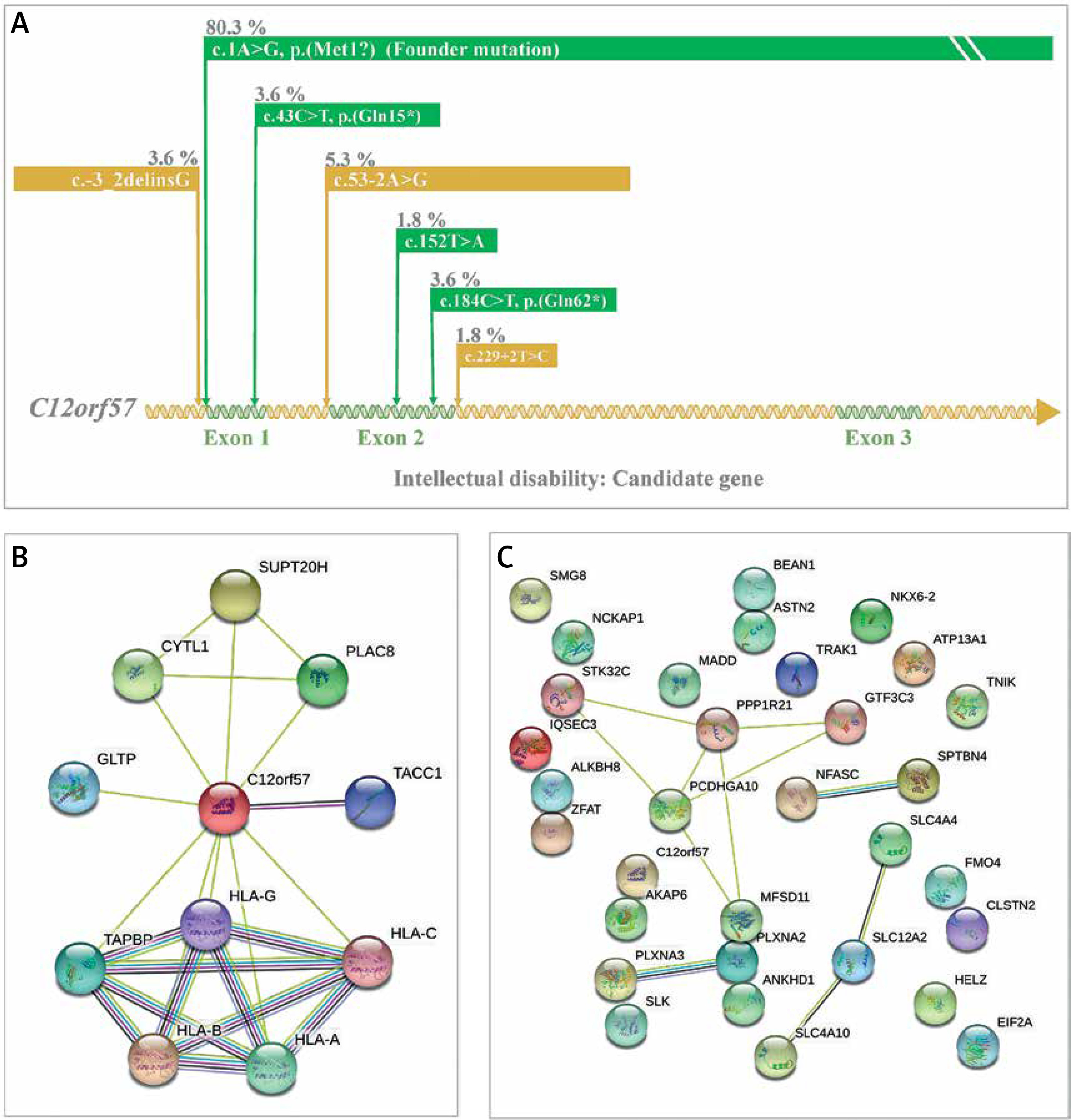
Intellectual disability is the most prevalent genetic disorder in terms of carrier frequency in the Saudi population (Figure 2) [7]. Temtamy syndrome is a type of intellectual impairment with epilepsy, ocular involvement and corpus callosum dysgenesis. A detailed study on temtamy syndrome exposed a high carrier frequency of start loss mutation [c.1A>G; p.M1?] in the chromosome 12 open reading frame 57 (C12orf57) gene in the Saudi population; it was the first observation of the syndrome for C12orf57. The study showed variable phenotypic in the cases with intellectual disability. The study conclusively reported the C12orf57 gene variants in the pathology of intellectual disability/developmental delay in the Arab population. Interestingly, the observations on the absence of typical syndromic features in Saudi ancestry are associated with intellectual disability, which is common and recessive. The start loss mutation [c.1A>G; p.M1?] in the C12orf57 gene is the most frequently prevalent (80.3% of all cases studied) disease marker in the Saudi ancestry (Figure 5) [69]. Another 6 variants in addition to start loss variation in the C12orf57 gene were reported (Figure 5) [69]. The seven variations in the C12orf57 gene are the founder mutation c.1A>G, p.(Met1?), c.53-2A>G, c.184C>T, p.(Gln62*), p.(Gln15*), c.229+2T>C, c.-3_2delinsG, c.43C>T, and c.152T>A, p.(Leu51Gln) accumulated in exons 1 and 2 (Figure 5) [69]. The C12orf57 gene was also associated with microphthalmia in the Saudi population (Table I) [66]. Large-scale exome sequencing and other studies reported the association between variations in AKAP6, ALKBH8, CLSTN2, EIF2A, IQSEC3, PLXNA2, PLXNA3, SMG8, BEAN1, HELZ, SLC4A4, SLC4A10 and TNIK and intellectual disability [70–75]. Intellectual disability patients (n = 105) from 68 families were identified with intellectual disability candidate genes such as ASTN2, ANKHD1, FMO4, ATP13A1, GTF3C3, MADD, NCKAP1, STK32C, NFASC, MFSD11, NKX6-2, PCDHGA10, SLC12A2, PPP1R21, SLK, SPTBN4, TRAK1, and ZFAT [76]. Expression analysis confirmed the intellectual disability candidate gene, NCKAP1 [76]. A study on Qatari subjects revealed the association of the novel pathogenic PGAP3 variant with hyperphosphatasia with mental retardation, global developmental delay and neuromuscular abnormalities and brain anomalies [45]. Protein-protein interaction analysis of the C12orf57 gene with the literature using STRING revealed the high interaction with HLA genes and associated pathways (Figure 5 B) [58]. Studies from Egyptians and Jordanians revealed the association of pathogenic SLC39A8, PQBP1, TTC5 and PUS3 variation in patients with intellectual disability [47, 48, 50, 51]. WES analysis from Lebanese subjects with intellectual disability identified the association of pathogenic variations in COQ8A and MED25 [77]. Further, the protein-protein interaction analysis (STRING [58]) of genes of intellectual disability candidate genes of the Arab population from various studies revealed (Figure 5 C) the most significant (p value < 0.05) biological processes, neuron differentiation (C12orf57, SPTBN4, TRAK1, NFASC, NCKAP1, PLXNA2, NKX6-2, PLXNA3, SLC4A10, and TNIK), neuron development (C12orf57, SPTBN4, TRAK1, NFASC, NCKAP1, PLXNA2, PLXNA3, SLC4A10, and TNIK), neuron projection morphogenesis (C12orf57, SPTBN4, TRAK1, NFASC, NCKAP1, PLXNA2, PLXNA3, and TNIK), central nervous system neuron differentiation (C12orf57, SPTBN4, NKX6-2, PLXNA3, and SLC4A10), central nervous system neuron development (C12orf57, SPTBN4, PLXNA3, and SLC4A10) and generation of neurons (C12orf57, SPTBN4, TRAK1, NFASC, NCKAP1, ASTN2, PLXNA2, NKX6-2, PLXNA3, SLC4A10, and TNIK).
Figure 5
A – Disease markers of intellectual disability in C12orf57 gene. Variants and their percentage contribution to the presence of intellectual disability in Saudi population. B – Protein-protein interaction analysis of C12orf57 gene with literature using STRING [58]. C – Protein-protein interaction analysis of genes of intellectual disability candidate genes of Arab population
Microphthalmia
Genetic investigation using multi-gene panel and WES on 93 families with developmental eye defect (microphthalmia) revealed 55 point variations in 24 disease genes, including 15 novel variations [66]. In addition, the study discovered interesting candidate variations in two genes, MYO10 and ZNF219, that have not been related to human disorders, which were notable novel possibilities for microphthalmia in the study, most of whom were Saudis, and there were 14 Egyptian and Lebanese subjects [66]. SLC18A2, PAX6, DSC3 and CNKSR1 genes are very rarely linked in the families with microphthalmia.
Metabolic traits
Despite concerted efforts at a national level to promote awareness about the dangers of a sedentary lifestyle and consuming fast food, the people in the Arab regions continue to be at risk for metabolic diseases. There are no well-known genetic risk factors for metabolic diseases in the Arab population like the European population, and the existing genetic risk loci for metabolic traits in the Arab population have not been shown adequately. A recent GWAS study on an Arab (Kuwaiti) population to identify genetic risk factors for quantitative characteristics including anthropometry, insulin resistance, lipid profile, and levels of blood pressure failed to discover associated recessive variations or were unable to replicate the identified loci [78]. This is mainly due to the complicated gene–environment interactions in Arab ancestries. However, another study on the same population identified the TNKS haplotype associated with hypertension [36].
Other genetic diseases
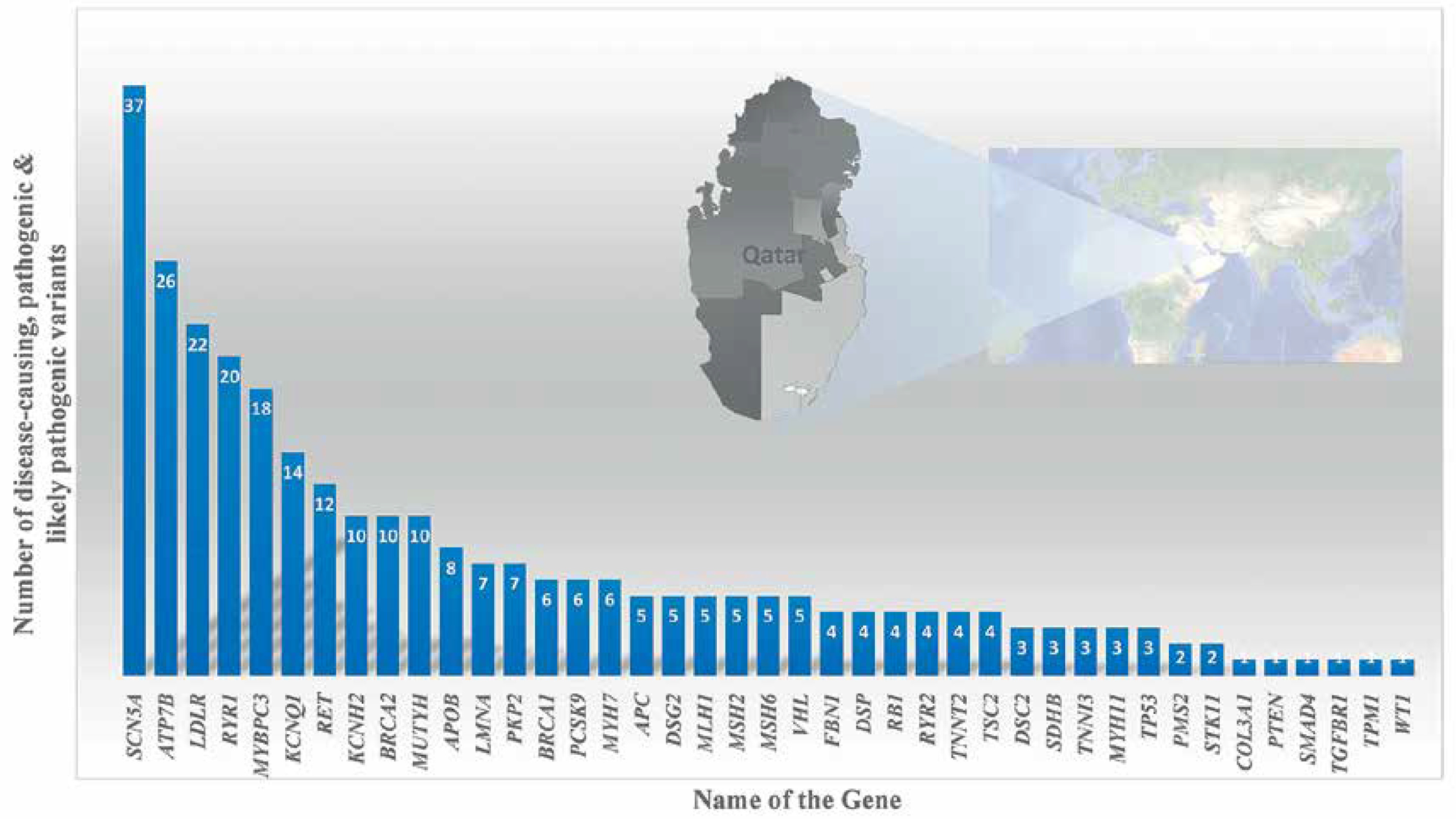
Variants in the Mendeliome in Saudi ancestry – APC-related Cenani-Lenz syndrome, Steel syndrome, syndromic cataract, oral-facial-digital syndrome, CHARGE-like presentation, epileptic encephalopathy, Ehlers-Danlos-like syndrome, and congenital hydrocephalus – were reported either with compatible phenotypes (homozygous variant in 30 genes) or phenotypes different from the original reports (homozygous mutations in 18 candidate genes) [79]. Studies on systemic juvenile idiopathic arthritis (LACC1 gene) [80, 81]; recurrent pregnancy loss (ASIC5 gene) [82]; tricho-hepato-enteric syndrome (SKIV2L and TTC37 genes) [83]; STING-associated vasculopathy of infantile-onset (STING1 gene) [84]; multiple congenital anomaly syndrome (SMG9 gene) [85]; diabetic retinopathy (NME3, LOC728699, and FASTK genes) [86]; congenital neutropenia with inflammatory bowel disease (G6PC3 gene) [87]; skeletal dysplasia (XYLT1) [88, 89]; Wolf–Hirschhorn syndrome (WHSC1 gene) [90]; lymphatic dysplasia with nonimmune hydrops fetalis (PIEZO1) [89]; Cohen syndrome (VPS13B gene) [91]; severe combined immunodeficiency disease (AK2, JAK3, and MTHFD1 genes) [92]; celiac disease (CPED1 gene) [93]; hereditary spherocytosis type 3 (SPTA1 gene) [89]; developmental delay, cerebellar hypoplasia, and myoclonic seizures (KCNMA1 gene) [74]; Cenani-Lenz syndrome (APC gene) [74]; Sjogren-Larsson syndrome (ELOVL4 gene) [74]; autism spectrum disorder (multigene) [94]; congenital heart disease (PRKD1 gene) [95]; ciliopathies [96]; Parkinsonism (PLA2G6 gene) [97]; retinal dystrophies (CLRN1, ABCA4, CERKL, AGBL5, CDH16, and DNAJC17 genes) [98–100]; pediatric asthma [101]; cardiovascular genetic diseases (LDLR gene) [102]; enteroendocrine dysfunction (PCSK1) [103]; tricho-hepato-enteric syndrome (TTC37 and SKIV2L) [104]; Wolcott–Rallison syndrome (EIF2AK3) [105], Fanconi–Bickel syndrome (SLC2A2) [105] and Alström syndrome (exon 19 skipping in ALMS1 gene) [106, 107] using whole exome or whole genome analysis revealed disease markers in the Saudi population. Families with autosomal recessive retinal dystrophies from various ethnicities including Saudis were analyzed for candidate genes using WES; 45 unique deleterious variants including 18 novel variants were observed [108]. UGCG (UDP-glucose ceramide glucosyltransferase) gene related ichthyosis was reported from a single Saudi family, caused by NM_003358:exon2:c.142dupA variations in the UGCG gene [109]. Epileptic encephalopathy in a consanguineous family was exome analyzed and a pathogenic variation in the FRRS1L [c.961C>T: p.(Gln321*)] gene was identified [110]. The NOTCH4 gene with a truncating and a splice variant was observed in 2 Saudi patients with Parkinson’s disease [111]. Furthermore, the study reported 18 genes (121 mutations) in Parkinson’s disease in 60 Saudis; most of the variants are missense (n = 90) and nonsense (n = 11) [111]. Gene specific pathogenic variant analysis of an Arab population of Qataris (n = 6045 whole-genome sequencing) revealed the most pathogenic and likely pathogenic variants in the SCN5A (n = 37 variants) gene followed by ATP7B (n = 26 variants), LDLR (n = 22 variants) and RYR1 (n = 20 variants) (Figure 6) [112]. The study revealed genes associated with Li-Fraumeni syndrome (TP53 gene); Peutz-Jeghers syndrome (STK11 gene); Lynch syndrome (MSH6, MLH1, PMS2, and MSH2 genes); familial adenomatous polyposis (APC gene); juvenile polyposis (SMAD4 and BMPR1A genes); Von Hippel–Lindau syndrome (VHL gene); multiple endocrine neoplasia type 1 (MEN1 gene); familial medullary thyroid cancer (RET gene); multiple endocrine neoplasia type 2 (RET gene); PTEN hamartoma tumor syndrome (PTEN gene); WT1-related Wilms tumor (WT1 gene); retinoblastoma (RB1 gene); hereditary paraganglioma pheochromocytoma syndrome (SDHAF2, SDHD, SDHB and SDHC genes); neurofibromatosis type 2 (NF2 gene); Loeys-Dietz syndromes, Marfan syndrome, and familial thoracic aortic aneurysms and dissections (TGFBR2, FBN1, ACTA2, TGFBR1, MYH11 and SMAD3 genes); tuberous sclerosis complex (TSC1 and TSC2 genes); Ehlers-Danlos syndrome (COL3A1 gene); Wilson disease (ATP7B gene); dilated cardiomyopathy, hypertrophic cardiomyopathy (MYH7, MYBPC3, TPM1, MYL3, TNNT2, TNNI3, LMNA, ACTC1, MYL2, PRKAG2 and GLA genes), arrhythmogenic right ventricular cardiomyopathy (DSC2, TMEM43, PKP2, DSP, and DSG2 genes); catecholaminergic polymorphic ventricular tachycardia (RYR2 gene); long QT and Brugada syndrome (SCN5A, KCNH2, and KCNQ1 genes); malignant hyperthermia susceptibility (RYR1 and CACNA1S genes); familial hypercholesterolemia (PCSK9, APOB, and LDLR genes); and ornithine transcarbamylase deficiency (OTC gene) diseases in an Arab population (n = 6045 Qataris, whole-genome sequencing) [112]. Low bone mineral density associated variants were found in non-coding RNA MALAT1 (antisense TALAM1), FASLG, SAG, LSAMP and FAM189A2 from a Qatari based Arab population with 3000 WGS [113] and the IL1RL1 gene variant with familial Mediterranean fever [114].
Figure 6
Number of disease-causing, pathogenic and likely pathogenic variants per gene through 6045 WGS of Qataris from QGP
Studies (WES) in Egyptian individuals revealed associated genes of primary hyperoxaluria type I (AGXT gene), infantile hypercalcemia/hypophosphatemia/nephrolithiasis (SLC34A1 gene) [115], severe combined immunodeficiency (JAK3 gene) [116], propionic acidemia (PCCA gene) [117], sulfite oxidase deficiency (SUOX gene), molybdenum cofactor deficiency (MOCS2 gene) [118], primary hereditary microcephaly (ASPM gene) [119], familial Mediterranean fever (MEFV gene) [120], thiamine-responsive megaloblastic anemia (SLC19A2 gene) [121], cerebellar atrophy and developmental delay (PLA2G6, KIF1A and MOCS2A genes) [122], micro-/anophthalmia (VSX2, SOX2, and FOXE3 genes) [123], RAG-deficiency (RAG1 and RAG2 genes) [124], auriculocondylar syndrome (PLCB4, GNAI3, and EDN1 genes) [125], ectodermal dysplasia (EDA, EDAR, and EDARADD genes) [126], 3-phosphoglycerate dehydrogenase deficiency (PHGDH gene) [127], Carpenter syndrome (RAB23 gene) [128], disorders/differences of sex development (NR5A1, CYP19A1, AMH, AMHR2, WT1, HHAT, and FANCA and in the X-linked genes KDM6A and ARX genes) [129] and autosomal recessive polycystic kidney disease (PKHD1 gene) [130]. WES studies in Iraqi people revealed associated genes of epileptic encephalopathy (SLC13A5 gene) [131], juvenile neuronal ceroid lipofuscinoses (CLN3 gene) [132], inherited thrombocytopenia (FYB gene) [133], microcephaly (TMX2 gene) [134], non-syndromic retinal dystrophy (POC1B gene) [135], progressive pseudorheumatoid dysplasia (WISP3 gene) [136], dedicator of cytokinesis 8 deficiency (DOCK8 gene) [137], and developmental delay (MED27 gene) [138]. A Jordanian patient with hypotrichosis-lymphedema-telangiectasia syndrome was analyzed using WES for causative genetic factors; the study revealed the SOX18 mutation associated with the syndrome [139]. WES analysis of four consanguineous Jordanian families detected the causative genetic variant responsible for an unknown gastrointestinal related diseases [140]. Across the five consanguineous families, four variations in the RP1 and RLBP1 genes were identified as disease-causing variants in patients with retinitis pigmentosa [141]. Two novel pathogenic variants in the DYSF gene were identified in the patients with muscular dystrophies from consanguineous Jordanian families [142]. Eight Jordanian consanguineous families with multiple keratoconic individuals were analyzed using WES, which revealed two variants in the genes MYOF and STX2, and one variant in the genes COL6A5, ZNF676 and ZNF765 [143]. WES analysis of Lebanese subjects with various genetic disorders (renal dysfunction, PKHD1, GRHPR and NPHS2 genes) revealed associated genes with the overall success rate of 56% [55]. Other studies on Lebanese with cytochrome c oxidase deficiency (PET100 gene) [144], discoid lupus erythematosus (TRAF3IP2 gene) [145], Bardet-Biedl and Usher syndromes (BBS9, ARL6, BBS12 and BBS5 genes) [146] and Basel-Vanagaite-Smirin-Yosef syndrome (MED25 gene) [147] revealed the causative genes. Genetic polymorphisms in Arab individuals with obesity were discussed previously [148–150].
Genetic analysis on Saudi patients (n = 98) with cholestatic liver disease revealed 37 variations including 15 novel mutations in ABCB11, ABCB4, ABCC2, AGL, ATP8B1, CYP7B1, FAH, TJP2, and VIPAS39 [67]. The ABCB11 gene was reported with the most common candidate disease marker gene among 25% of cases studied [67]. In Saudi patients (n = 261) with primary immunodeficiency an NGS sequencing platform through a targeted gene panel (162 PID genes) revealed 89 mutations (45 mutations and 44 novels) [151]. Sickle cell disease is the 5th most prevalent genetic disorder in terms of carrier frequency in the Saudi population [7]. High fetal hemoglobin (HbF) in sickle cell patients is common among Saudis; whole genome studies may reveal candidate HbF-modulating genes and ‘missing heritability’ in the Arab ancestries. WES based Trio (n = 16) analysis of attention deficit hyperactivity disorder (ADHD) reported 32 rare variants in 31 candidate genes; exclusively 5 genes (PSRC1, PTP4A3, NEK4, NLE1, and TMEM183A) were first observed among the Saudi ancestry [152]. Genomic studies on congenital microcephaly proposed BPTF, CCNH, MAP1B, and PPFIBP1 genes as candidate genes and confirmed ANKLE2, THG1L, YARS, and FRMD4A genes as congenital microcephaly candidate genes [153]. Targeted sequencing on Saudi patients (14 families) with Glanzmann thrombasthenia discovered 17 variants in ITGA2B and ITGB3 genes including 6 novel mutations [154]. The largest Arab study on 80 patients from 72 families (mostly consanguineous) with peroxisomal disorders (molecularly characterized) identified disease-causing variants (n = 43; 50% novel) and estimated the disease burden (~1 : 30,000) [155]. Exome sequencing and immunophenotyping of CARMIL2-deficient T cells confirmed the candidature of a novel 13bp frameshift deletion (c.2536_2548del) and substitution p.R50T in the CARMIL2 gene for combined immunodeficiencies [156].
Mitogenome
Sequencing and analysis of the entire mitochondrial DNA (mtDNA) genomes of Emirate females (n = 232) and comparison with African ancestries were carried out to characterize the genetic landscape of the Arab population from United Arab Emirates [157]. The mitochondrial genome of the Arab (Emirati) population from the United Arab Emirates revealed the prevalence of haplogroups, heteroplasmy, genetic variation, and demography. The study revealed 968 variations in the mtDNA genome in 15 haplogroups. Another study on the mitogenome of the Kuwaiti population (288 whole-exome) identified 1,241 mtDNA SNPs [158]. In a Qatari (n = 864) population mitogenome analysis of 1831 mtDNA variants including 56 indels and 1775 SNPs was performed [159]. A report from the Emirati population showed that the Arab population received sufficient gene flow from African ancestries. Furthermore, the study showed that the Arab population received sufficient gene flow from Africa ancestries and a demographic bottleneck was observed that occurred around the time of Western European contact [157]. Since the earliest modern humans emerged from Africa, multiple maternal lineage migratory events favoring African-Asian corridors have occurred, according to the research based on UAE mitogenomes [157]. Ancestral sharing with Africa, the Near East and East Asia was also revealed from the study [157]. Complete mitochondrial haplogroup analysis in Arab populations from Qatar and Kuwait based investigations revealed the relationship of mtDNA variations with obesity [158, 159]. The missense variant identified in the MT-ND2 gene, MT:5460G > A, in the Kuwaiti population is the most significant mtDNA variation associated with obesity [158].
Summary and future directions
Even though the KGP, SHGP, QGP, BGP and EGP are revisiting the genetics and genomics of Arab populations’ ancestries, lack of complete coordination between the initiatives is a major limitation on revealing the real disease markers of the Arab population [6]. Recent genomic studies identified more than 3000 new nucleotide variations linked to more than 1200 uncommon genetic diseases and numerous pathogenic variants in HGMD and the ClinVar Database have been reclassified as benign. However, more root level studies are needed on validating these changes from pathogenic to benign using Sanger sequencing and expression levels in the Arab population. More attention is needed on levels of intellectual impairment as they are the highest among Saudi Arabians from consanguineous families. Large-scale genomic research has opened up a fresh perspective on Mendelian genes and disorders. Arab research institutions must make a concerted effort to develop better strategies for doing more large-scale genotype-phenotype association studies in hereditary disorders prevalent in the region. Whole genome sequencing in large-scale control subjects including patients with inherited diseases can discover the molecular basis in the unsolved cases in the Arabian studies. Transcriptome studies in the Saudi population shall reveal the functional and molecular impact on the disease development by the candidate genes and pathogenic variations. Novel variants observed in inherited diseases will be the best resource for the developing disease marker panel for the Arab ancestry. Complicated gene–environment interactions in Arab ancestries with various diseases will open real networking factors on complex diseases prevalent in the Arab population. National coordination between regional research centers in the country and in the Arab region can reveal in-depth gene networks in diseases and associated pathways.