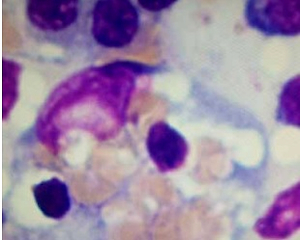
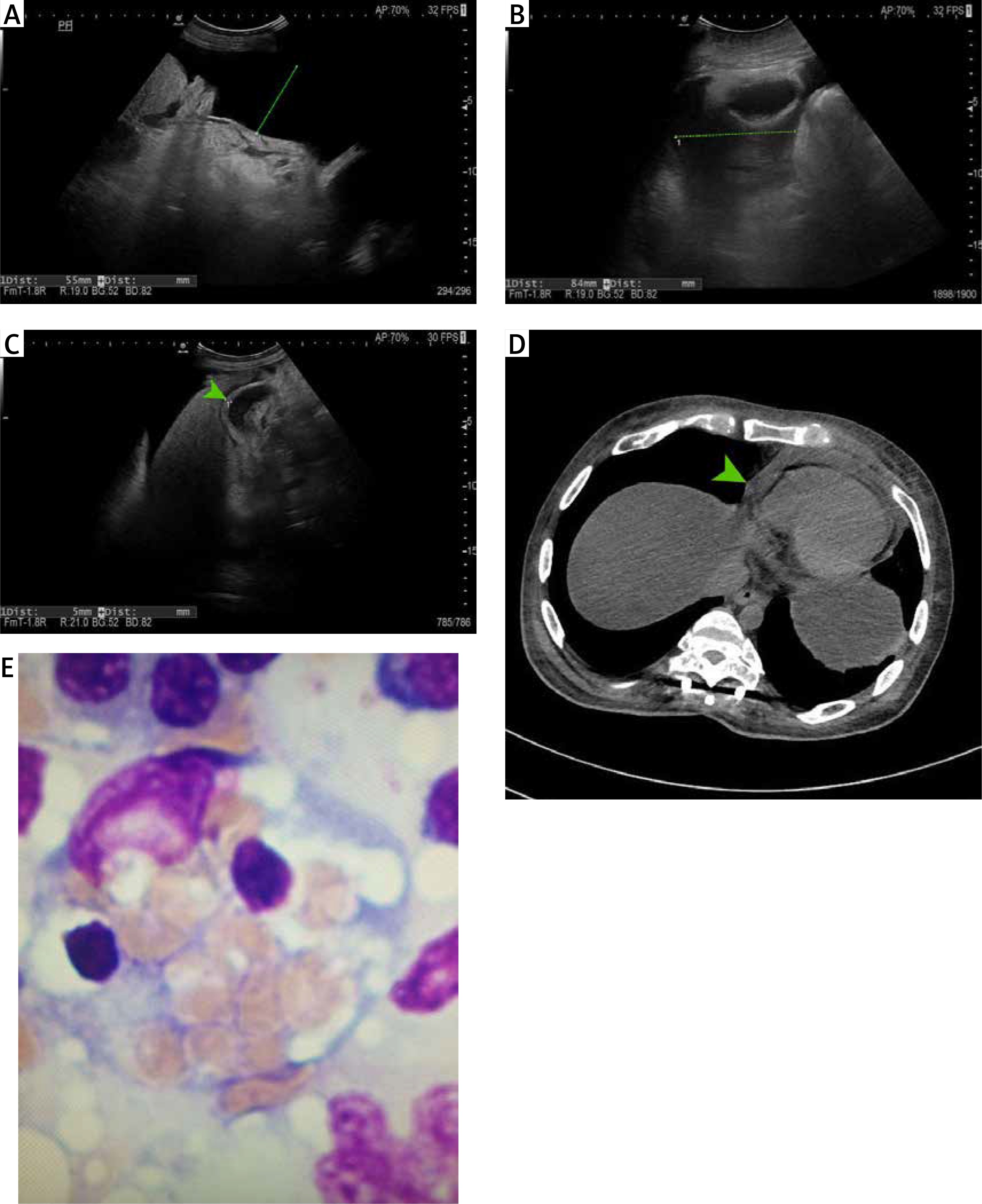
A 26-year-old male with achondroplasia presented with an 8-month history of recurrent fever, peripheral oedema, and ascites. Physical examination showed brown discoloration around the umbilicus, papular and keratotic lesions on the skin of the back, tachycardia, jaundice, ascites, and peripheral oedema. Laboratory findings included leukopaenia (3670/μl), increased activity of liver enzymes (alanine aminotransferase: 274 U/l, aspartate aminotransferase: 488 U/l, alkaline phosphatase 812 U/l, γ-glutamyl transferase 389 U/l), hyperbilirubinaemia (3.41 mg/dl), hypoproteinaemia (4.84 g/dl), hypoalbuminaemia (1.84 g/dl), prolonged activated partial thromboplastin time (aPTT 60 s), decreased fibrinogen concentration (133 mg/dl), increased concentration of dimer-D (13078 ng/ml) and substantially elevated ferritin (19973 ng/ml). Autoimmune work-up was negative. Serological investigations showed no active Epstein-Barr virus (EBV), but IgG antibodies were found. Blood cultures grew Staphylococcus epidermidis and Staphylococcus hominis, and urine cultures grew (ESBL+) Escherichia coli. Abdominal ultrasonography revealed ascites (Figures 1 A, B), hepatic steatosis, and a thick-walled gallbladder with biliary sludge (Figure 1 C). High-resolution computed tomography (HRCT) revealed pericardial effusion (pericardium thickness of 12 mm) (Figure 1 D). Initial treatment including culture-directed antibiotics and intravenous loop-diuretic resulted in clinical improvement with significant reduction of peripheral oedema and ascites. Clinical signs and prior laboratory findings with further progression of pancytopenia, considered together, suggested hemophagocytic lymphohistiocytosis (HLH). Primary HLH was excluded after a bone marrow biopsy assessed by 2 independent haematologists was negative for hemophagocytes. Secondary HLH with prior sepsis as the initiatory factor was treated with a course of steroids, with good clinical response. However, the condition of the patient deteriorated within 30 days of discharge with intermittent hyperpyrexia, massive ascites, tachycardia, and hypotension. Laboratory workup showed persistent cytopaenia, and elevated ferritin and C-reactive protein. Blood cultures and tests for infective endocarditis were negative. Due to the signs of urinary tract infection, corticosteroid treatment was discontinued and broad-spectrum antibiotics as well as granulocyte colony-stimulating factor were administrated. Despite the applied treatment the patient remained feverish. Because of the advanced caries being a potential cause of recurrent infections, the patient was transferred to the Maxillo-Facial Surgery Department and underwent surgical removal of the teeth. Immediately afterward, the patient was transferred to the Department of Haematology for further diagnostics. Trepanobiopsy revealed the presence of hemophagocytes (approx. 4%). The patient met 5 out of 8 diagnostic criteria for HLH [1], with an H score 80–88%.
Figure 1
A – Abdominal ultrasound showing the features of ascites – the thickness of the fluid layer is up to 55 mm in the right flank (the green dotted line shows the place where the thickness was measured). B – Abdominal ultrasound showing the features of ascites – the thickness of the fluid layer in the pelvis is 84 mm (the green dotted line shows where the thickness was measured). C – Abdominal ultrasound revealing a thickened wall of the gallbladder (the green arrow indicates the gallbladder wall). D – High-resolution computed tomography (HRCT) showing pericardial effusion (the green arrow indicates the pericardial effusion). E – Bone marrow trepanobiopsy shows hemophagocyte
After completion of the HLH 2004 treatment protocol our patient showed marked clinical improvement, with no further hyperpyrexia, recovery of cell counts, and a decline in inflammatory markers and ferritin.
HLH is a syndrome of uncontrolled hyperinflammation, which, if untreated, leads to death [2]; however, as a rare condition, it is largely underdiagnosed [3]. Therefore, a timely diagnosis and introduction of adequate treatment is of utmost importance.