Introduction
Thymomas and thymic carcinoma (TC) are rare thymic epithelial tumours (TETs), mostly localised in the anterior mediastinum [1]. Thymomas are not frequent tumours, having an annual incidence of 0.15 patients per 100,000 person-years [2, 3]. Here we consider thymomas that have been represented as a very indolent form of rare tumours, but with the ability for local and regional invasion. They do not exhibit usual cell atypia, and they are frequently associated with paraneoplastic syndromes, including the most common association – myasthenia gravis [4]. According to WHO classification, thymomas are divided into five subcategories: A, AB, B1, B2, and B3 considering cell morphology, degree of atypia, and differentiation of epithelial cells and lymphocytes [5]. Many attempts have been made to identify target genes for successful therapy of TETs. Still, genomic analysis, with the emphasis on disease rarity, which caused a limited number of studies, has revealed several genes related to this pathology, such as EGFR, HER2, KIT, KRAS, and T53 [6].
Next-generation sequencing (NGS) is a powerful tool for sequencing large numbers of samples, which covers the whole genome, the whole exome, or targets genes of interest, enabling large datasets for various types of conditions, such as cancer. This kind of disease arises from germline mutations and/or somatic mutation accumulation during a lifetime, mostly in a context of specific transcriptome profile and epigenetics factors [7]. So far, only a few studies have been published using a high-throughput approach explaining the molecular landscape of thymoma. Here we analysed 35 thymoma patients using targeted NGS methodology by TruSeq Amplicon Cancer Panel (TSACP) consisting of 48 solid tumour-related genes frequently involved in tumourigenesis.
Material and methods
Patients
The study was approved by the Ethics Committee of the Clinical Centre of Serbia. The study included a cohort of 35 patients with thymoma. All the diagnoses of thymoma were established using histopathological (PH) tissue sample preparations obtained either by surgery procedure or by fine-needle aspiration biopsy (FNAB), where the surgery was not possible to perform. Thymoma were classified according to World Health Organisation (WHO) and the Masaoka-Koga stage classification. Additionally, 35 healthy control samples were analysed.
DNA isolation and library preparation
Genomic DNA from 35 thymoma patients was isolated from formalin-fixed, paraffin-embedded (FFPE) tumour tissue and from blood of 35 healthy controls (Qiagen, Valencia, USA). The library preparation wasperformed using 250 ng of genomic DNA, according to the manufacturer’s protocol (Illumina Inc., San Diego, CA, USA).
TruSeq Amplicon – Cancer Panel
TruSeq Amplicon – Cancer Panel, TSACP (Illumina Inc., San Diego, CA, USA) targets mutational hotspots in 48 cancer-related genes (ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAQ, GNAS, HNF1A, HRAS, IDH1, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MLP, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, and VHL), TSACP consists of 212 amplicons captured by pairs of oligonucleotides designed to hybridise flanking targeted regions of interest. The TSACP platform that we used was designed to detect exclusively low-frequency somatic variants in a mixed cell population (bellow 5%), in the most frequently mutated specific hotspot regions, which were highly specifically targeted in the most common mutated oncogenes associated with solid tumours.
TruSight One Sequencing Panel
TruSight One Sequencing Panel probes were built to cover the main exons, targeting in total a 12 Mb region size. This panel comprises the exons within 4813 genes. The probes were designed to ensure optimal performance and depth coverage, minimum 20× and maximum 100×, on 95% of chosen regions.
Bioinformatics analysis
A FASTQ file was generated by removing low-quality bases (base quality lower than 30) using quality trimming of 3’ ends of non-index reads with low-quality score. This step was performed during the alignment procedure using BWA (Burrows Wheeler Aligner). The alignment process of read sequences to the reference genome hg19 generated BAM (Binary Alignment /Map) file using a BWA-SW aligner (an extended version of – Smith-Waterman, adapted for sequence reads longer than 70 bp). The final step was a variant calling procedure, which detected single nucleotide polymorphisms (SNPs) and insertions/deletions written in Variant Call Format (VCF). For the generation of a final output file VCF we used GATK (Genome Analysis Toolkit, www.broadinstitute/gatk), a Somatic Variant Caller developed by Illumina. The Illumina-developed Somatic Variant Caller in somatic mode was chosen for variant calling. In order to identify and interpret disease-relevant variants from the Variant Call Format (VCF) we used Illumina VariantStudio. Moreover, for variant annotation we used an additional computational tool: Variant Effect Predictor, VEP [8]. The Integrated Genomics Viewer [9] was used for visual evaluation of the data. For summarising genomic alterations within the oncogenes across the group of patients, we used the OncoPrinter [10] tool. For mutation mapping on a linear protein and its domains we used cBioPortal tool Mutation Mapper [11].
Statistical analysis
Survival probabilities were estimated using the Kaplan-Mayer method, and differences in the survival distributions were evaluated using log-rank test [12]. Statistical analyses were performed using SPSS computer software, version 21.0 (IBM Corp). For all analyses, the p-values were two-tailed, and p < 0.05 was considered statistically significant.
Results
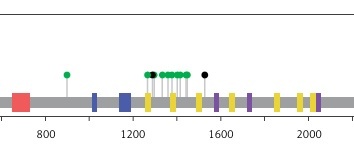
In the present study, we used targeted next-generation sequencing (NGS) to analyse 1.225 × 106 bp sequences from 35 thymoma patients. The TruSeq Cancer Panel (TSACP) was used for somatic variant detection in specific genomic regions, including 212 amplicons in 48 important cancer-related genes. The average coverage was 145× per targeted sequences, with 75,786 total reads, with median read depth of 595×. The total number of detected variants in present study were 4447, out of which 2906 in coding region (median per patient 83 with a range: 2–300) and 1541 in the non-coding area (median per patient 44 with a range: 0–172) (Figure 1 A). For further analysis, we only considered variants that potentially modulate protein structure and function, including nonsense (N), frameshift (F), and missense (M) changes (NFM). In all patients, we identified 1963 potential protein modulating variants with a median per patient of 56, in a range from 2 to 195 variants. Eight patients emerged with the highest number of variants, harbouring more than 100 NFM variants: #20 (PH tip B3), #28 (PH tip B1), #34 (PH tip AB), #44 (PH tip A), #53 (PH tip B3), #59 (PH tip B1), #61 (PH tip AB), and #62 (PH tip A), among them patient #20 with 195 variants (Figure 1 B). Additionally, we identified 276 splice variants (Figure 1 C). PH type B thymoma was present in 50% of patients, but observing subtypes, we had almost equal distribution of subtypes among the cases: 2 patients were detected by each subtype A, AB, B1, and B3, while B2 was not present in this group of patients.
Figure 1
The number of mutations per thymoma patient. A – Total number of mutations in coding and non-coding regions identified by targeted NGS. B – Distribution of nonsense (N), frameshift (F), and missense (M) mutations in coding regions of targeted genes. C – Distribution of splice variants per patient
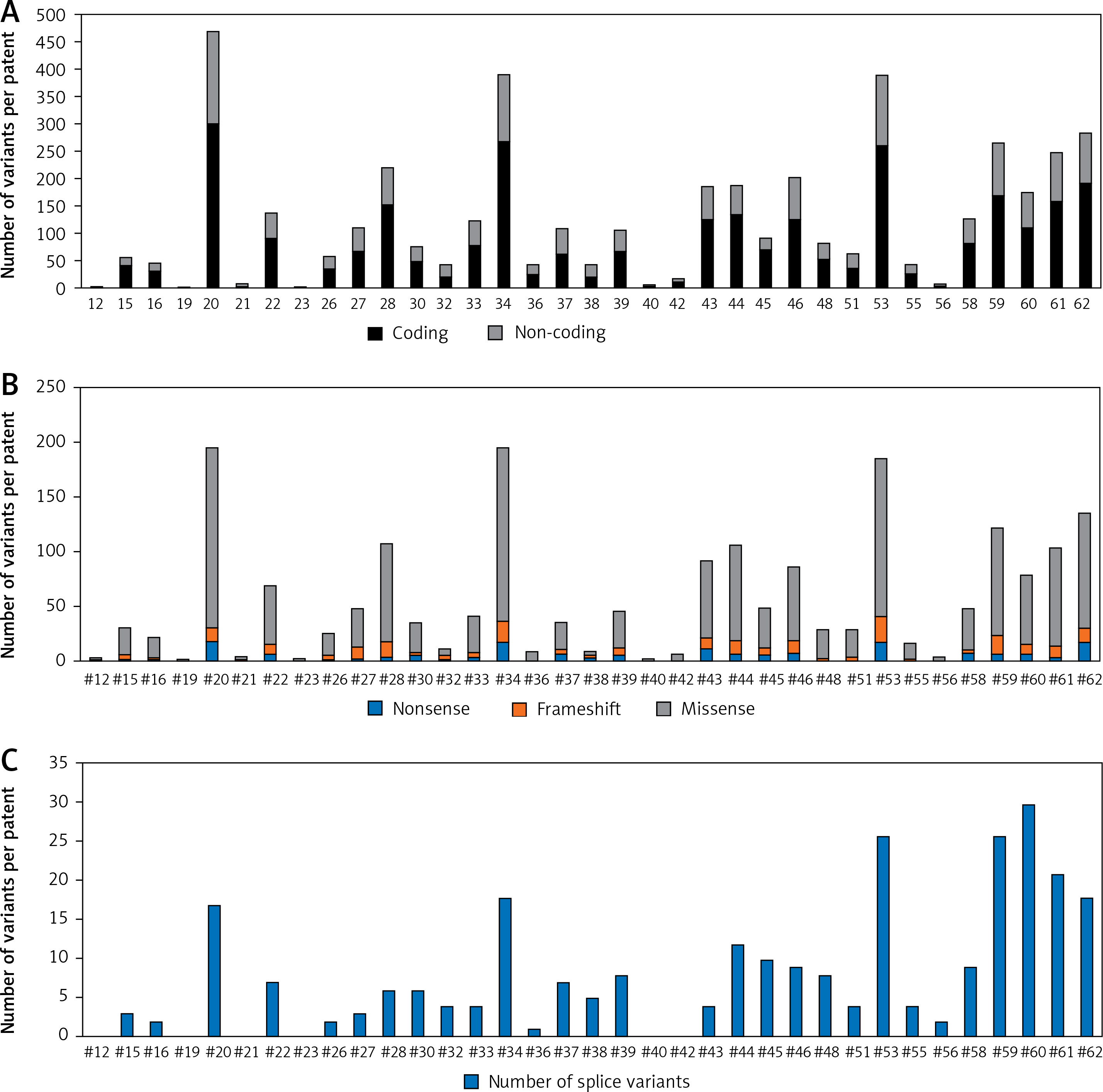
Four genes appeared as the highest mutated genes, including APC, ATM, ERBB4, and SMAD4, having more than 100 NFM variants, indicating a potential contribution of these genes in thymoma pathogenesis. Additionally, EGFR, FBXW7, FGFR3, FGFR2, GNAQ, GNA11, HNF1A, KIT, MET, PIK3CA, PTEN, and RB1 were highly mutated harbouring more than 40 NFM changes. TP53 and KDR contained more than 90 NFM variants, out of which the majority were well-known polymorphisms (rs1042522 and rs1870377). On the other hand, some of the genes selected using the TSACP panel including FGFR1, MPL, NPM, and SRC had less than five NFM variants (Figure 2).
Figure 2
The number of mutations per targeted gene. Distribution of NFM mutations in the coding regions of targeted genes
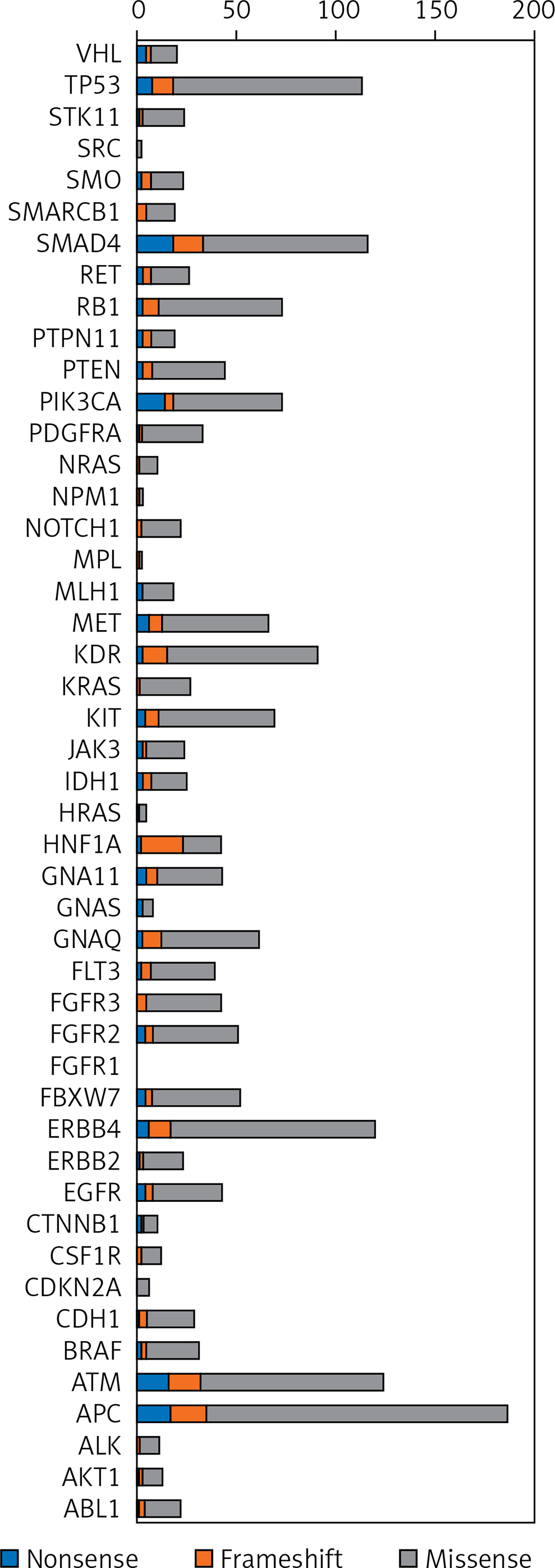
Protein-changing variants in SMAD4, APC, ATM, PTEN, KDR, and TP53 were present in more than 70% of analysed cases. The distribution of somatic variant types in 48 selected cancer-related genes across the patients is represented in OncoPrint (Figure 3).
Figure 3
OncoPrint showing the distribution of genetic alterations in 48 targeted tumour suppressor and oncogenes in 35 thymoma patients. The type of mutations are labelled in the colour legend, particular genes in rows, and tumour samples in columns
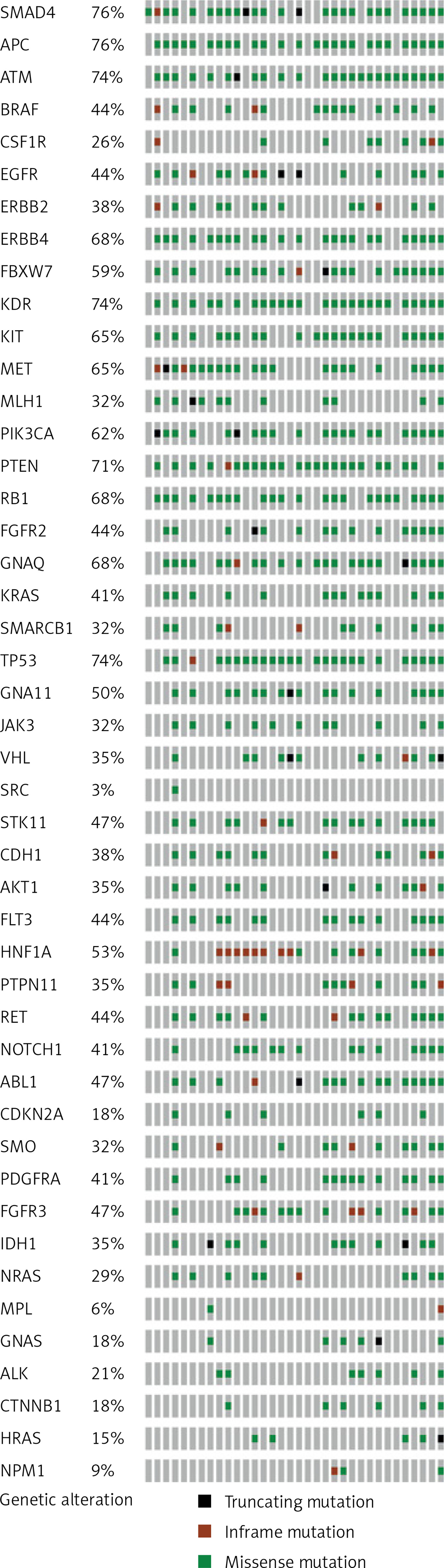
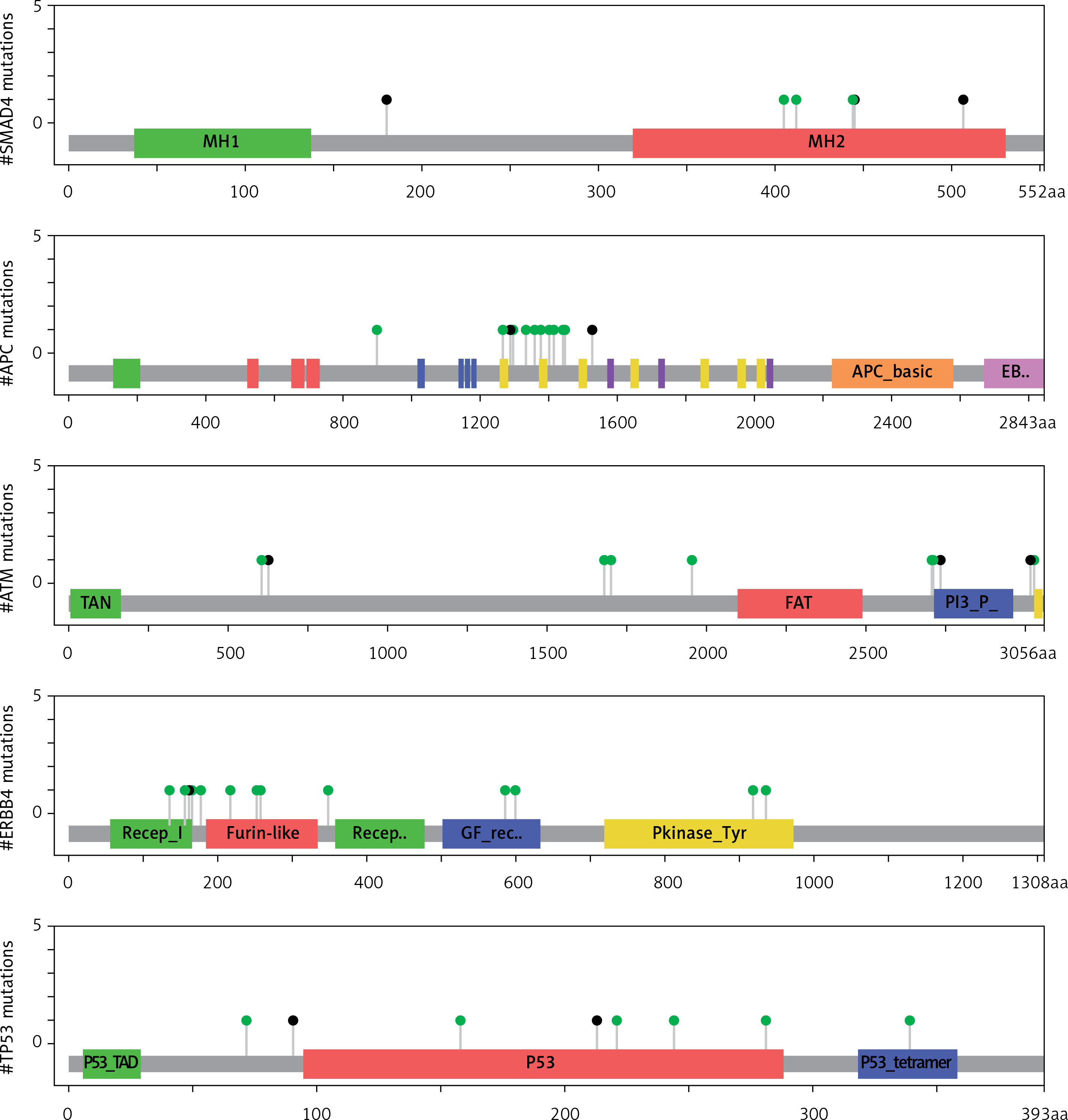
Moreover, this study revealed 168 recurrent variants, which were present at least in two samples in the analysed cohort of patients, out of which 25 were stop gained, nine frameshift, and 134 substitution variants. The recurrent variants within the genes with the highest number of NFM variants are presented with map mutations on linear protein and its domain (lollipop plots) (Figure 4).
Figure 4
The protein sequence and functional domains are depicted on the x-axis. The number of SMAD4, APC, ATM, ERBB4, and TP53 variants is depicted on the y-axis. Green circles correspond to missense variants. Black circles correspond to nonsense variants
For further analysis, we selected previously reported variants with pathogenic effect. Variant pathogenicity was confirmed using SIFT and PolyPhen, and was checked in the dbSNP, COSMIC, OncoKB, ICGC Data Portal, and ClinVar databases. The pathogenic variants and their functional impact are presented in Table I.
Table I
Pathogenic variants in thymoma patients
Additionally, we analysed three samples from healthy individuals using an amplicon-based TSACP platform, which includes pathogenic somatic variants detected in this study. None of the somatic variants found in thymoma patients were detected in controls. Moreover, we analysed 32 healthy control samples using exome sequencing covering pathogenic variants identified in this study (TruSight One Platform, Illumina). Using this approach, which detects germline variants, we did not identify any variant found in thymoma patients. Our findings point out that the variants we reported in this study are somatic thymoma-specific variants.
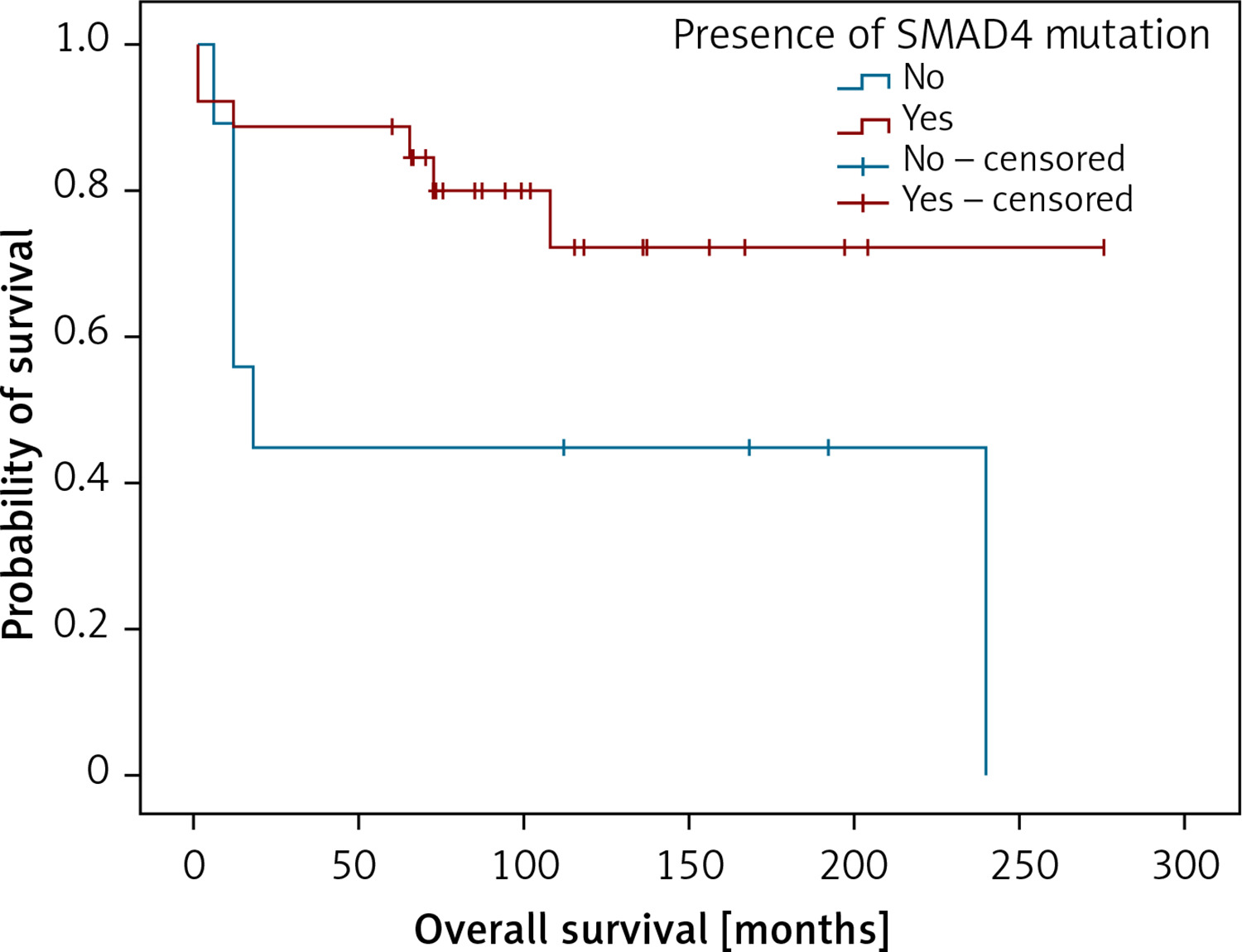
Also, analysing genetic findings and clinical data we were able to establish that the presence of variants in SMAD4 gene predicted shorter overall survival in the patients (Figure 5).
Figure 5
Kaplan-Meier analysis of overall survival in respect to the presence of damaging SMAD4 variants. Patients with SMAD4 mutation (n = 26; 20 censored), patients not containing SMAD4 mutations (n = 9; 3 censored). LogRank test (χ2 = 4.022; p = 0.045), positive vs. negative group – 231 vs. 113 months
Discussion
Advances in next-generation sequencing (NGS) of tumour genome have enabled the discovery of novel variants, included single variants (SNVs), small insertions/deletions (indels), copy number variations (CNVs), and translocations, related to tumourigenesis. Identification of genomic alterations using NGS leads to the discovery of new biomarkers in order to improve diagnosis, prognosis, and, finally, treatment of various cancer types [7]. The most comprehensive high-throughput approach, whole-genome sequencing (WGS), enables assessment of all potentially damaging changes in the genome. The disadvantages of this methodology are high cost per sample and low read depth, which limits their potential to detect SNVs with low allele frequencies (less than 5%), and the time needed for sequencing and data analysis. Consequently, targeted NGS, which targets hotspot regions or specific genes of interest, has been frequently applied. Targeted NGS provides high coverage, which is necessary for detection of rare variants in heterogeneous tumours or in low-purity samples [13, 14]. Our aim was to study somatic variants in the thymoma genome. We used the TSACP, a previously validated targeted gene panel [15, 16]. For variant calling we used a somatic caller, which detects somatic variants, and the coverage that we obtained is sufficiently high to confirm the presence of somatic variants. The comparison of tumour samples with matched control of non-tumour samples of each patient is very important, in order to eliminate false positive and germline variants. The limitation of our study is that the control non-tumour samples, for example, a buccal swab of each patient, were not available. Instead of this, we included three samples from healthy individuals analysed on the TSACP platform and 32 samples analysed on clinical exome sequencing. Because we did not detect in any control sample variants considered to be either somatic (TSACP) or germline (TSO panel), we presume that the variants identified in this study are somatic thymoma-specific variants.
We found four genes, namely: APC, ATM, ERBB4, and SMAD4, having more than 100 protein-changing variants. Particular variants in these genes were recurrent (appearing in at least two patients). Moreover, EGFR, FBXW7, FGFR3, FGFR2, GNAQ, GNA11, HNF1A, KIT, MET, PIK3CA, PTEN, and RB1 genes were highly mutated harbouring more than 40 NFM variants, while TP53 had 90 NFM changes including familiar polymorphism rs1042522. Due to the rarity of the pathology that we investigated, only a few studies concerning high-throughput sequencing datasets related with thymoma have been previously published. Also, the molecular pathology of this disease has been poorly understood, particularly regarding A, AB, B1, B2, and B3 subtypes. In a previously published study by Belani et al., in which whole genome and whole exome approaches on one cytogenetically normal B3 patient were used, the authors suggested that DNMT3A (p.G728D) and ASXL1 (p.E657fs) variants are involved in thymoma genesis [17]. Petrini et al., using whole genome and transcriptome sequencing, identified one translocation t(11;X), copy number gain of chromosome 1q, 5, 7 and X and CN loss of 3p, 6, 11q42.2-q and q13, 10 SNVs, and two indels, suggesting the need for additional screening for better understanding of disease genetics [15].The Ekner group analysed two B3 thymoma using an Ion AmpliSeq Cancer Hotspot Panel, and found a mutation in noncoding regions of the SMARCB1 and STK11 gene and non-synonymous HRAS mutation in A thymomas [16].
The most frequently mutated genes detected in this study belong to EGFR signalling, ATM, TP53 signalling pathways, regulating cell cycle check points, gene expression, and apoptosis. Analysing the results of the present study, we found that the SMAD4 variants were present in 27 cases. Three of the SMAD4 variants, SMAD4 (Q180*, R445*, K507*), were nonsense recurrent variants with probable oncogenic potential according to OncoKB [18], suggesting their potential contribution to thymoma pathogenesis. Moreover, clinical results in our study demonstrated a statistically significant difference in cumulative survival between patients with the presence of damaging variants in SMAD4 and the patients without these variants. Previously published results suggested that pathogenic variants in SMAD4 gene in these tumours were related with poor prognosis of the disease [19]. Some results using a quantitative proteomic approach pointed out SMAD4 as a potential candidate gene with a specific role in the pathogenesis of thymoma [20].
Additionally, we detected pathogenic variants in the following genes: APC (C1289*, Q1529*), ATM (Q628*, Q2733*), TP53 (D281N, G244S, R158C, E221K, R213*, W91*), PTEN (R233Q, KRAS V14I), and RET C634Y, potential targets for drug therapy and drug testing, which could lead a step closer to better treatment and disease outcome. The pathogenic effect of variants was confirmed using SIFT and PolyPhen, and was checked in the dbSNP, COSMIC, OncoKB, ICGC Data Portal, and ClinVar databases.
In this work, we found that the APC gene was mutated in 27 patients. Among the variants that we identified, C1289* and Q1529* emerged as pathogenic variants in this oncogene. APC was characterised as a key tumour suppressor factor involved in several fundamental cellular processes including tumourigenesis and homeostasis, especially of epithelial cells and lymphocytes [21]. In addition to APC, the most frequent genetic variations in all types of thymomas are present in 6p21.3 (MHC locus) and 6q25.2–25.3. But, so-called “common gatekeeper” or common tumour suppressor genes that could be involved in key processes in thymic progenitor cells have not yet been identified. The alterations in APC gene locus have been associated with the aggressive subtypes B2 and B3. “High-risk alteration” at APC locus was found in AB type, which are not aggressive forms of the disease, suggesting the existence of another tumour suppressor gene that has the opposite effect to APC [22].
The ATM gene encodes for the phosphatidylinositol 3-kinase, with the main responsibility of repairing double-stranded DNA breaks. Next-generation sequencing analysis pointed out that the ATM gene as one of the most aberrant genes in solid and haematological tumours. The COSMIC database harbours 167 distinct variants related to various tumours, excluding unknown alterations [23]. In our work, ATM was mutated in 26 patients, having two nonsense variants (Q628*, and Q2733*) with a pathogenic effect on the clinical phenotype.
TP53 has been described previously as the candidate gene related to disease pathogenesis [6]. Non-synonymous mutations in this gene were detected in highly aggressive forms of the disease [16]. TP53 and BCOR were the most frequently mutated genes in TCA and B3 thymomas, respectively [19]. We identified two TP53 variants (R213* and V91*) with stop codon producing truncated protein probably with damaging function, and four pathogenic missense variants (D281N, G244S, R158C, E221K).
Additionally, we found the KDR gene, encoding for tyrosine protein kinase, which had variants in 74% of cases with one nonsense recurrent variant and eight missense recurrent variants. Moreover, the PTEN gene, which has missense variants with various oncogenic level, were present in 71% of patients.
The most recent studies on thymoma and other pathologies like glioblastomas and lung adenocarcinoma are related to transriptomic profiling [24–28]. Nevertheless, there amplicon-based technology that we used for thymoma analysis provides reliable and usable data for optimal treatment options, due to its high coverage detection of low-frequency somatic variants. Small-targeted panels with high coverage have already been accepted for translational research, molecular diagnostics, and targeted therapy [29, 30]. Our results could contribute to design of thymoma-specific small-targeted panel, applicable in clinical practice. Moreover, cell lines harboring pathogenic variants detected in this study would be a valuable source for drug testing.
In conclusion, a big challenge in precision medicine, imply identification and distinction between rare driver variants, responsible for disease development, and passenger’s mutations, which have no impact on tumour phenotype. All somatic variants within the hotspot regions of the observed genes that we identified contribute both to the knowledge of molecular pathogenesis of the thymoma and to the understanding of the clinical phenotypes of the patients. However, further high-throughput screening is needed to complement and improve current knowledge and provide insight into this rare pathology.