Introduction
Hearing loss is a universal auditory dysfunction. Severe hearing loss strongly impacts human daily communication and social activities. Based on the latest WHO data in 2019, there are about 466 million hearing-impaired people worldwide, accounting for 5% of the world’s total population [1]. The Second National Sample Survey on Disabilities in 2006 revealed that there were 27.8 million people with hearing disabilities in China, of whom 20.04 million were simply people with hearing disabilities, accounting for 24.2% of the total [2]. Neonatal deafness incidence is 1–3‰, that is, about 30,000 deaf children are born every year. The China Birth Defects Prevention Report (2012) pointed out that hearing disabilities have become the second largest birth defect in China. Therefore, preventing hearing disabilities and intervening in birth defects are essential aspects to improve the quality of the Chinese population. Studies have shown that at least 60% of cases of severe hearing loss result from genetic factors [3]. Hereditary hearing loss mainly involves four types: autosomal recessive inheritance (approximately 80%), autosomal dominant inheritance (approximately 15–20%), mitochondrial inheritance (approximately 1%) and sex-linked inheritance (approximately 1%) [4]. Based on a molecular epidemiological survey of deafness in China, GJB2, GJB3, SLC26A4 and mitochondrial 12S rRNA are the most universal deafness genes in Chinese [5, 6]. The hereditary hearing loss resulting from GJB2 and SLC26A4 follows autosomal recessive inheritance. Carriers of mitochondrial 12S rRNA variants are sensitive to aminoglycoside drugs, belonging to maternal inheritance, whose deafness rate in Chinese with hearing loss is up to 34% [5]. However, survey data reveal that among the normal population in China, about 6% are carriers of common deafness gene variants [7, 8], and most deaf newborns each year are born to families without a family history of deafness. It can be seen that although deafness gene screening and intervention for people with hearing loss can prevent birth of deaf children by deaf parents to a certain extent, it cannot effectively prevent normal parents from giving birth to children with hereditary hearing loss. Therefore, screening of common deafness genes in the normal population before the marriage check or pregnancy and early gestation, early detection of couples with pathogenic variants in the same gene and corresponding intervention measures are effective means to prevent hearing disabilities.
In this study, we conducted common deafness gene screening in 60,391 women of childbearing age in Weihai, genetic counseling in high-risk families and corresponding intervention measures, which can effectively prevent these families from giving birth to children with hearing loss, thus reducing the incidence in the region and providing genetic counseling and clinical decision making from the genetic screening.
Material and methods
Research subjects
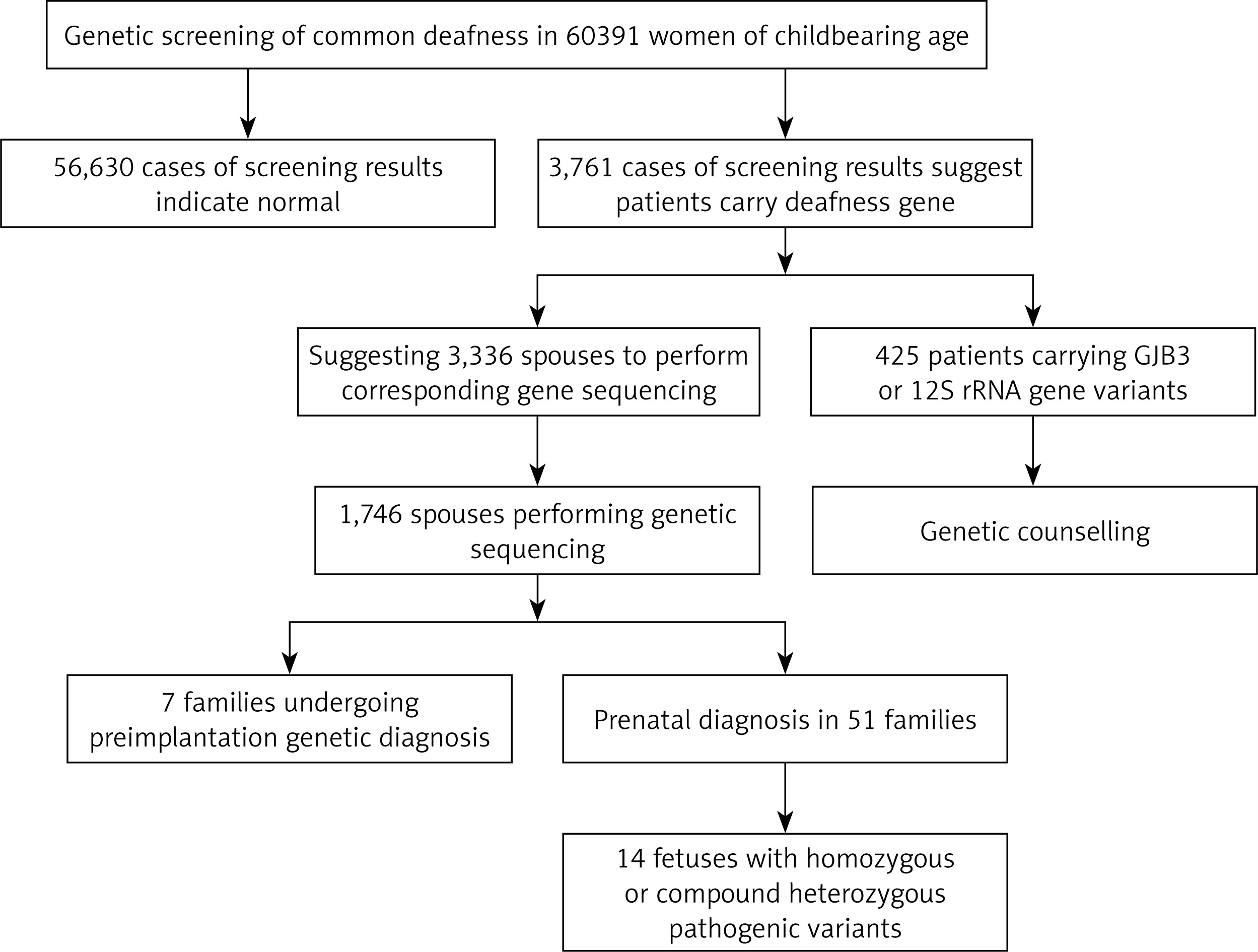
In total 60,391 pre-pregnancy/early-pregnancy women who received treatment in second-level or above hospitals in Weihai between February 2017 and December 2019 were selected, ranging in age from 18 to 47, and gestational age less than 15 + 6 weeks. Among them, there were in total 53 women with different degrees of hearing loss, and 17 with family history of hearing disabilities. This study was approved by the Medical Ethics Committee of Weihai Maternal and Child Health Hospital, and all research subjects signed informed consent forms.
All patients completed the survey by answering questions and filling in the “deafness questionnaire survey form.” The details were as follows: (a) basic information: name, gender, date of birth, nationality, marital status, contact information, etc.; (b) morbidity status: age of onset, presence of other associated symptoms (vertigo, tinnitus, etc.), related medical history (infectious diseases, etc.), history of use of aminoglycoside antibiotics, long-term noise exposure, otitis media or traumatic history of the ear, etc.; (c) the development condition of language, whether wearing a hearing aid or cochlea, etc.; and (d) whether other systemic diseases co-existed (eyes, bones, intellectual disability, etc.). Ear examination and audiological evaluations including pure-tone audiometry, immittance testing, auditory brainstem response, and auditory steady-state response were performed for all hearing impaired patients. Physical and neurological examinations were carried out with special attention to renal and ophthalmologic differences to exclude those with syndromic deafness (Figure 1).
DNA extraction
Venous or peripheral blood of our subjects was collected to prepare dry blood slices on filter papers (Whatman 903), each with at least 2 blood spots. An automatic punching machine was used to take a slice of dried blood sample to corresponding 96-well plates complemented with reagents, and it was sealed with aluminium film. The plates were placed on a heater to boil, and cooled for later use. QIAamp DNA Blood Mini Kit (Qiagen, Germany) was used to extract genomic DNA from each blood sample following the manufacturer’s instructions, and a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA) was used to evaluate the quantity and quality of extracted DNA.
Library construction
Twenty common variant sites of GJB2, SLC26A4, and GJB3, as well as mtDNA, were detected by using a deafness diagnostic screening panel (CapitalBio Genomics Co., Ltd., China). Library construction, quality control, and sequencing template preparation were performed according to the instructions of a Blood DNA LQ kit. The PCR method was used to amplify 20 variant sites in GJB2, GJB3, SLC26A4 and mitochondrial 12S rRNA genes, then sequencing labels were added and purified to complete library construction.
Deafness gene detection
Via the high-throughput sequencing method and gene sequencer BGISEQ-500 to detect deafness genes, 20 pathogenic sites in 4 genes were detected, including in GJB2 c.35delG, c.167delT, c.176_191del16, c.235delC and c.299_300delAT, GJB3 gene c.538C>T and c.547G>A, in SLC26A4 c.281C>T, c.589G>A, c.919-2A>G, c.1174A>T, c.1226G>A, c.1229C>T, c.1707+5G>A, c.1975G>C, c.2027T> A, c.2162C>T and c.2168A>G, and in mitochondrial 12S rRNA m.1494C>T and m.1555A>G.
For women who have been screened for the pathogenic sites in deafness genes, it is recommended that their spouses undergo corresponding gene sequencing, providing medication guidance for women with drug-sensitive gene variants. For couples who carry pathogenic variants in the same gene, genetic counseling was provided and corresponding intervention measures were taken. Postpartum neonatal hearing screening and follow-up for women with deafness gene mutations were done.
Results
Deafness gene screening results of 60,391 women
Among 60,391 pre-pregnancy/early-pregnancy women, in total 3,761 women carried common deafness gene variants, with a total carrier rate of 6.23% (3,761/60,391) (Table I), 1724 (2.85%) carried only one GJB2 variant, 1,534 (2.54%) only carried one SLC26A4 variant, 300 (0.5%) only carried one GJB3 variant, and 125 (0.2%) carried the drug-sensitive gene variant. There were 44 women (0.07%) carrying two deafness gene variants, of whom 38 carried both GJB2 and SLC26A4 variants, accounting for 0.06% of the total screened population, 3 carried both GJB2 and GJB3 variants, and 3 carried SLC26A4 and GJB3 variants at the same time. Additionally, there were 53 women with clinical manifestations of varying degrees of hearing loss, of whom 15 (0.02%) carried 2 pathogenic variants in GJB2 at the same time, 19 (0.03%) carried 2 pathogenic variants in SLC26A4 at the same time, 3 carried gene variants for drug-sensitive deafness, 4 only carried 1 variant in GJB2 or SLC26A4, and 12 did not carry common variants.
Table I
Common deafness gene screening results of 60,391 cases
GJB2. In this study, 1,780 women carried the GJB2 variant, with a carrying rate of 2.92% (1,780/60,391). Among them, the carrying rate of c.235delC was the highest at 2.08%; c.299_300delAT was the second with 0.73%; the carrying rate of c.35delG was the lowest at 0.005% (Table II).
Table II
Statistics of GJB2 variant types
SLC26A4. In total 1,594 women carried the SLC26A4 variant, with a carrying rate of 2.62% (1,594/60,391). Among them, the carrying rate of c.919-2A>G was the highest at 1.63%, followed by c.2168A>G, with a carrying rate of 0.42% (Table III).
Table III
Statistics of SLC26A4 variant types
Deafness gene sequencing results of spouses
Based on the female test findings, we recommend that the husbands of 3,336 women with GJB2 or SLC26A4 variants undergo corresponding gene sequencing. In total 1,746 women’s spouses agreed to be tested. The detection rate was 52.3% (1746/3336) and 139 men carried GJB2 or SLC26A4 variants, with a carrying rate of 7.96% (139/1,746). Among them, 1,146 men were sequenced for GJB2; 77 were carrying known pathogenic variants, and 5 carrying suspected pathogenic variants or variants of unknown clinical significance. In total 600 men were sequenced for SLC26A4; 31 were carrying known pathogenic variants, and 26 carrying suspected variants or variants of unknown clinical significance. From the results, the known pathogenic variants were mainly c.109G>A (2.18%), c.235delC (1.54%), c.299_300delAT (0.52%) and c.919-2A> in SLC26A4 and G (0.98%) in GJB2. At the same time, in SLC26A4 suspected pathogenic variants and variants of unknown clinical significance accounted for a relatively high proportion of 1.49% (Table IV).
Table IV
Deafness gene variant of 1,746 spouses
Results of fetus receiving prenatal diagnosis
Based on the clinical manifestations and test results of couples, it was finally determined that 99 couples carried known or suspected pathogenic variants in GJB2 or SLC26A4, and their offspring have a 25% risk of disease, which is in line with the prenatal indications for diagnosis. But among these 99 couples, 38 husbands carried the GJB2 c.109G>A variant. At present, although the GJB2 c.109G>A variant is considered to be a pathogenic variant, the hearing phenotype resulting from it is quite different: hearing can be normal or weakened, and it also can be manifested at birth or later, so if the offspring is diagnosed as a GJB2 c.109G>A homozygous variant or compound heterozygous variant before birth, their hearing phenotype after birth cannot be predicted. Finally, after genetic counseling and full informed consent, in total 51 couples underwent prenatal diagnosis, including 6 with the GJB2 c.109G>A variant (Table V), and 7 chose to undergo pre-implantation genetic diagnosis. The results of prenatal diagnosis showed that the offspring of 14 couples inherited deafness gene variants other than GJB2 c.109G>A from their parents. It is most likely that they are hereditary deafness patients, accounting for 27.5%. The offspring of 2 couples were a GJB2 c.235delC/c.109G>A compound heterozygous variant and a c.109G>A/c.299_300delAT compound heterozygous variant. The offspring of 23 couples may carry only one deaf gene variant or they did not inherit variants of their parents, and the predicted risk of the fetus with hereditary deafness is extremely low.
Table V
Prenatal diagnosis results of 51 couples
Among 125 women carrying drug-sensitive gene variants, 97 carried 12S rRNA 1555A>G homogeneous variants, 21 carried 12S rRNA 1555A>G heterogeneous variants, and 7 carried 12S rRNA 1494C>T homogeneous variants (Table I), respectively, accounting for 0.16%, 0.03% and 0.01% of the total people screened. The follow-up of women and their maternal family members revealed 3 women with hearing disability and 1 uncle with hearing disability. Because drug-susceptibility gene mutations follow the maternal inheritance method, we issued medication guide cards to 125 carriers of drug-susceptibility gene mutations, informing them and their maternal family members that they should absolutely avoid using aminoglycoside drugs throughout their lives in case of deafness.
Follow-up results of the offspring’s hearing
Our work conducted follow-up of the offspring’s hearing of 3,761 female carriers; 21 carriers’ children were finally diagnosed with hearing disabilities.
Among them, 18 children were tested for deafness genes; 12 carried two variants in GJB2 or SLC26A4, and they were clearly children with congenital hereditary hearing loss; 6 did not carry or only carried one variant in GJB2 or SLC26A4 (Table VI).
Table VI
Confirmed children with hearing and deafness gene test results
Discussion
The GJB2 variant is the first cause of hereditary nonsyndromic hearing loss (NSHL) among Chinese. At present, there are more than 100 gene variants in GJB2 that are known to be pathogenic (https://hereditaryhearingloss.org.), and these variants have obvious regional and ethnic differences [9]. There are also differences in common GJB2 variant sites in Chinese from different regions [10]. SLC26A4 is the second most universal variant gene in NSHL patients, and patients of different races also have different gene variant profiles [11–13]. In China, c.919-2A>G is the most common variant [14]. Based on previous studies, in our hospital the main pathogenic variant of NSHL patients in the Jiaodong area is GJB2 c.235delC, followed by SLC26A4 c.919-2A>G [15]. Among 60,391 women of childbearing age in this study, 1,739 only carried the GJB2 variant, with a carrying rate of 2.88%; 1,553 women only carried the SLC26A4 variant, with a carrying rate of 2.58%. The carrying rates of GJB2 and SLC26A4 variants among Chinese were 3% and 2–3%, respectively, which are basically the same [7, 8]. Among them, GJB2 c.235delC has the highest carrying rate, accounting for 2.08%, followed by SLC26A4 c.919-2A>G, with a carrying rate of 1.63%, which further confirms that common deafness gene variants in Jiaodong are mainly GJB2 c.235delC and SLC26A4 c. 919-2A>G.
GJB2 encodes a gap junction protein (connexin 26 protein – Cx26) [16]. The most common pathogenic variant in GJB2 among Chinese is c.235delC, if which lack its base will result in frameshift variants in the coding region of GJB2, leading to premature termination of translation and the formation of non-functional Cx26, which results in sensorineural hearing loss [17]. The GJB2 c.235delC homozygous variant is mostly manifested as congenital severe-extremely severe hearing loss, and very few cases are delayed moderate post-lingual hearing loss [18]. Chang et al. [19] conducted audiometric phenotype statistics on 100 NSHL patients with the GJB2 c.235delC homozygous variant, finding that patients with deafness caused by the GJB2 c.235delC homozygous variant showed diversity in both ear hearing phenotypes and the degree of hearing loss is mainly extremely severe, severe and moderately severe, whereas mild is the least common. In this study, 6 women were detected as GJB2 c.235delC homozygous variants, and their hearing phenotypes were also heterogeneous, including 4 women with congenital extremely severe hearing loss in both ears, 1 with moderate hearing loss, and 1 with mild hearing loss. This has accumulated original data for us to further exploit the mechanism of different hearing phenotypes of patients carrying the same gene variants, thereby providing a possible basis for treating genes in NSHL patients.
In 2013, Pu et al. [20] proposed a tertiary prevention strategy for hearing disabilities of birth defects based on genetic screening and diagnosis: First, via drug-induced deafness susceptibility screening, fertility guidance for deaf couples, early-pregnancy general screening, pre-love guidance for young deaf people to achieve primary prevention; second, through prenatal diagnosis and intervention to achieve the secondary prevention; third, through neonatal gene screening to achieve tertiary prevention. At present, the joint screening of neonatal hearing and deafness genes is being widely promoted in China and has produced certain findings. A number of studies [21, 22] have shown that hearing combined with deafness gene screening is an effective means for early detection of late-onset hereditary hearing loss and drug-sensitive individuals. It provides opportunities for early treatment and lingual rehabilitation for children, with important clinical significance.
In this study, 60,391 women of childbearing age were screened for common deafness genes. With the actual detection rate of spouses being 52.3%, 99 high-risk families were finally identified. After genetic counseling, 7 families opted for preimplantation genetic diagnosis, and 51 families for prenatal diagnosis. The findings of prenatal diagnosis showed that 14 fetuses were most likely to have hearing loss, and the diagnosis rate was 27.5% (14/51). It can be seen that carrying out common deafness genetic screening among women of childbearing age and providing follow-up services such as genetic counseling for high-risk families is essential for reducing birth of children with hearing disabilities.
In our practice of screening common deafness genes in women of childbearing age in this region, we have summarized the following experiences:
Common deafness gene screening is highly accepted among women of childbearing age in this region. On average, more than 90% of pre-pregnancy/early-pregnancy women are voluntarily tested each year. In addition to publicity and education, government departments also played an important role in including the testing project in people’s livelihood to implement free testing. It can be seen that the government departments provide strong support for preventing and controlling birth defects in terms of policies and funds, which effectively promotes the prevention and control of birth defects in the region.
The rate of further testing for the spouses of female carriers is low, 52.3%, which is far lower than that of females, especially in SLC26A4 sequencing, only 37.6% (600/1594). After analysis, the first reason is that most families believe that the hearing of both parents is normal, with no family history of hearing disabilities, and that the probability that both couples carry the same type of deaf gene variant is very low. Second, the cost for spouses to receive corresponding gene sequencing is relatively high and needs to be borne by themselves. If these families were highly compliant with our recommendation at that time, through genetic counseling and prenatal diagnosis, we could at least predict the hearing status of the offspring in 47.6% (10/21) of families and provide them with the opportunity to choose. Therefore, how to improve the general people’s correct understanding of hereditary deafness is a problem that we urgently need to solve.
Be cautious about genetic counseling of controversial pathogenic variants. The detection rate of the GJB2 c.109G>A variant is relatively high in both normal people and patients with hearing disabilities. Existing research statistics show that c.109G>A is a pathogenic variant, but the hearing phenotypes caused by it are quite different: hearing can be normal or weakened, and the degree of hearing loss can range from mild to severe. This study finally determined that in families where both couples carry known pathogenic variants in the GJB2 gene, 38 husbands are carriers of c.109G>A. With full knowledge, 6 families chose prenatal diagnosis, and finally 2 families had fetuses with the c.109G>A compound heterozygous variant (1 case was the c.109G>A/c.299_300delAT compound heterozygous variant; 1 case was the c.235delC/c.109G>A compound heterozygous variant). We followed up the hearing of their offspring, and both families indicated that their offspring had passed the newborn hearing screening. Because the later clinical manifestations of this group of people are unpredictable, we recommend that the two families undergo regular hearing monitoring to accurately assess their hearing level.
Although hearing impairment is not a serious disability or mortal disease, it will seriously affect the quality of life and inflict a heavy economic and psychological burden on families. In this study, prenatal diagnosis was applied to predict that 14 fetuses were likely to have hearing loss. We learned from follow-up that 12 of them chose to terminate the pregnancy and suggested that these families should give priority to preimplantation genetics if they have reproductive needs, with diagnosis to prevent the occurrence of hereditary deafness. Therefore, with continuously advancing society and generally improved awareness, realizing primary prevention of birth defects of hearing disabilities, that is, to carry out genetic screening for hereditary deafness before pregnancy or even before marriage, and actively adopt corresponding intervention measures, will more effectively avoid various economic and social ethical issues.
In conclusion, screening for common deafness genes in women of childbearing age and providing follow-up consultation and diagnosis are effective means to prevent birth deafness. Such a screening mode is available to achieve early diagnosis and intervention of hereditary deafness, providing families with an opportunity to make informed choices. It can also warn those carrying drug-sensitive deafness genes to avoid using ototoxic drugs in case of hearing loss. But it is still necessary to further promote the publicity and education of deafness gene screening, popularizing the related knowledge of hereditary deafness, especially to strengthen genetic counseling for high-risk families, so that they have a clearer understanding of hereditary deafness, are more willing to receive deafness gene testing, and further understand the importance of preimplantation genetic diagnosis or prenatal diagnosis to prevent hereditary deafness. Additionally, the government’s strong support for preventing and controlling birth defects will also encourage more people to receive testing to a certain extent.