Introduction
Ovarian torsion results from rotation of the ovarian vascular pedicle and leads to obstruction of arterial inflow and venous outflow [1]. The common first-line treatment is conservative management, which includes detorsion of the torsioned segments [2]. The main purpose of treating ischemia is to improve tissue perfusion by re-establishing blood flow. Maintaining circulation and reperfusion following a period of ischemia creates a new pathophysiological process called reperfusion injury, which causes tissue damage to various degrees. Oxidative stress plays a pivotal role in I/R injury in ovarian tissue [3] and reperfusion could be the main cause of the oxidative stress injury [4].
Tumor necrosis factor-alpha (TNF-α) is an important cytokine which participates in many inflammation-related processes [5]. TNF-α receptors play a decisive role in inflammation and can be found on the cell membrane and in plasma in a soluble form. Etanercept is a fusion protein of the extracellular domain, and consists of the p75 TNF receptor and the Fc domain of human immunoglobulin G1 [6]. Etanercept administration following endotoxin injection is reported to neutralize the activity of TNF-α and inhibit release of cytokines, chemokines, and stress hormones [7].
In view of these findings, we aimed to investigate how etanercept would affect ovarian ischemia/reperfusion (I/R) injury as well as ovarian reserve. We evaluated histopathological changes and ovarian follicle count as well as serum anti-Mullerian hormone (AMH) levels and tissue levels of glutathione (GSH), malondialdehyde (MDA), superoxide dismutase (SOD) and myeloperoxidase (MPO) to evaluate oxidative stress, and 8-hydroxydeoxyguanosine (8-OHdG) and caspase-3 activity for DNA injury and apoptosis determination in rats exposed to ovarian I/R damage.
Material and methods
This study was conducted at the Acıbadem University Experimental Animal Laboratory, Istanbul, Turkey. Experimental procedures and protocols were approved by the Local Animal Ethics Committee of Acıbadem University (Approval Number: 2014/26). All protocols were performed in accordance with the National Health and Medical Research Council guidelines for the care of experimental animals. This study was reported in accordance with ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines [8]. Rats were randomly assigned to standard cages with two animals per cage, and maintained in the laboratory under controlled environmental conditions on a 12-h light/dark cycle with a room temperature of 21 ±1°C, and also had free access to water and food.
Following determination of the estrus stage for each animal with daily vaginal smears, the rats were randomly divided into four groups: sham (control) group (n = 6); sham + etanercept group (n = 6) (intravenous 10 mg/kg etanercept 30 min before laparotomy); ischemia/reperfusion (I/R) group (n = 6); I/R + etanercept group (n = 6) (intravenous 10 mg/kg etanercept 30 min before laparotomy + I/R).
Rats were sedated with 60 mg/kg of 10% ketamine hydrochloride (Ketasol; Richter Pharma, Weis, Austria) and 10 mg/kg of 2% xylazine (Rompun; Bayer Health Care, Whippany, NJ, USA). Before the operation, a 1 ml blood sample was obtained from the rat’s jugular vein while under anesthesia. The abdominal skin was shaved and disinfected with 10% povidone iodine solution and the abdominal cavity was accessed through a 3 cm midline lower incision. In the sham group, the operation was ended after observation of the ovaries for 1 min. In the sham + etanercept group, 10 mg/kg etanercept was administered intravenously 30 min before the operation, which ended after observation of the ovaries for 1 min.
In the I/R group, ischemia was induced for 3 h by twisting the ovary, including the tuba-ovarian vessels, through one complete turn in a clockwise direction and clamping the pedicle with a 20–25 gram-per-square-centimeter pressure bulldog clamp (Vascu-Statt, Scanlan, St. Paul, MN, USA). We chose a 3-h duration of torsion based on the study of Ozler et al. [3]. The 3-h phase of ischemia was followed by a 24-h period of reperfusion (detorsion). Following detorsion, the abdominal incision was closed in two layers. Etanercept was administered 30 min before the operation to the I/R + etanercept group (10 mg/kg, intravenous) and ovarian I/R was performed with the same method.
In order to decrease the number of animals in the research study, in addition to the pre- and postoperative blood samples, 24 ovaries of 12 similar rats that were used in our previous study (Approval Number 2014/22; same ethical committee) were used again as histopathologic specimens for the I/R and sham groups.
At the end of the reperfusion period, 1 ml blood samples were obtained from the rat’s jugular vein for measurement of post-reperfusion AMH levels. The rats were euthanized and the bilateral ovarian tissue was harvested. The ovarian tissue was vertically divided into halves: one-half of the ovary was put into a 10% neutral-buffered formalin solution for 24 h for histologic examination. The other half was cleaned of retroperitoneal white adipose tissue and rapidly stored in a –80ºC freezer until required for biochemical analysis. All collected blood samples were immediately centrifuged for 10 min at 5000 rpm at +4°C. The serum obtained was transferred into Eppendorf tubes and stored at –80ºC until assayed.
Histopathologic evaluation
After fixation, samples were embedded in paraffin wax. Sections were cut from the paraffin blocks at 5 μm using a Leica RM2125RTS (Leica Biosystems, Nussloch, Germany) and stained with hematoxylin & eosin. All slides were examined under a light microscope (Olympus BX-50, Olympus Corp, Tokyo, Japan).
The criteria for ovarian injury were determined as interstitial edema, dilatation, hemorrhage, polymorphonuclear leukocyte infiltration (PNL), and follicular cell degeneration (granulosa cells). For every sample, criteria were scored from 0 to 3 (0 = none; 1 = mild; 2 = moderate; 3 = severe) [9]. The histological technique for follicle count was derived from the work of Durlinger et al. [10]. Follicles were classified in groups based on mean diameter: primordial (< 20 μm), preantral (20–220 μm), small antral (221–310 μm), and large antral (311–370 μm) [11]. Ovarian slides were examined by the same pathologist, who had been blinded to the test groups.
Biochemical analysis
Measurement of serum AMH levels
All collected blood samples were centrifuged at 2500 g and 4°C for 10 min, and sera were separated and stored at –80°C until assayed. Serum concentrations of AMH were quantified using an enzyme-linked immunosorbent assay (ELISA) anti-Mullerian Hormone Kit according to the manufacturer’s instructions (Bioassay Technology, Shanghai). The lower limit of sensitivity of the AMH ELISA was 0.051 ng/ml. The degree of precision of the ELISA system in terms of intra-assay and inter-assay coefficients of variance were < 10% and < 12%, respectively
Measurement of tissue myeloperoxidase activity
MPO activity was measured in ovarian tissue with the method of Hillegass et al. [12]. Tissue samples were homogenized with a potassium buffer (20 mM KH2PO4, pH 7.4) containing 0.5% (w/v) hexadecyltrimethylammonium bromide. We assessed its activity by measuring H2O2-dependent oxidation of o-dianisidine dihydrochloride. One unit of enzyme activity was measured at 460 nm and results were expressed as U/mg protein.
Measurement of tissue malondialdehyde and glutathione levels
Ovarian samples were homogenized in 150 mmol/l KCl for the determination of MDA and GSH levels. MDA levels were assayed for products of lipid peroxidation by monitoring thiobarbituric acid reactive-substance formation, as previously described [13]. Protein concentrations were measured by Bradford reagent and lipid peroxidation was expressed as nmol MDA/mg protein. GSH measurements were performed using metaphosphoric acid (Na2HPO4) for protein precipitation, using the Ellman procedure [14]. In brief, after centrifugation at 2000 g for 10 min, 0.5 ml of supernatant was added to 2 ml of 0.3 mol/l NaH2PO4 solution. Then 0.2 ml of dithiobisnitrobenzoate (0.4 mg/ml 1% sodium citrate) was added and absorbance at 412 nm was measured immediately after mixing. Protein concentrations were measured by Bradford reagent and GSH levels were expressed in μmol GSH/mg protein.
Measurement of superoxide dismutase activity
The SOD activity in ovarian tissue samples was measured in accordance with the method of Mylroie [15]. Briefly, measurements were performed in cuvettes containing 2.8 ml of 50 mM potassium phosphate (pH = 7.8) with 0.1 mM EDTA, 0.1 mM 0.39 mM riboflavin in 10 Mm potassium phosphate (pH 7.5), 0.1 ml of 6 mM o-dianisidine 2 HCl in deionized water, and 0.1 ml of tissue extract. Cuvettes with all their components were illuminated with 20-W Slylvania Grow Lux fluorescent tubes that were placed 5 cm above and to one side of cuvettes maintaining a temperature of 37°C. A standard curve was prepared routinely with bovine SOD (Sigma Chemical Co, S-2515-3000 U) as a reference. Absorbance was measured at 460 nm. Protein concentrations were measured by Bradford reagent and results were expressed as U/mg protein.
Measurement of tissue 8-hydroxy-2′-deoxyguanosine levels
Tissue samples were collected and genomic DNA was immediately extracted from the tissue using a commercial DNA extraction kit (PureLink) in accordance with the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). Samples were stored at –80°C prior to determination of 8-OHdG. Measurement of tissue 8-OHdG levels was performed using a competitive ELISA with the OxiSelect Oxidative DNA Damage ELISA kit as described in the manufacturer’s instructions (Cell Biolabs, San Diego, CA, USA).
Measurement of caspase-3 activity levels
The caspase-3 activity assay was performed using a caspase-3 cellular activity assay kit (Calbiochem, San Diego, CA, USA) according to the manufacturer’s instructions. Tissue samples were treated for 10 min with ice cold lysis buffer supplied by the manufacturer. Then 40 μl of tissue sample and 50 μl of assay buffer were added to each well and incubated at 37°C for 10 min. The colorimetric release of p-nitroaniline (pNA) from the Ac-DEVD-pNA substrate was measured at 450 nm. Protein concentrations were measured by Bradford reagent and caspase-3 activity was calculated as pmol/min/mg protein.
Western blot analysis for caspase-3
Caspase-3 protein expression was also measured directly by Western blot. Protein concentrations in homogenized samples were determined by Bradford reagent. Afterwards, 40 μg of protein was resolved on 12% SDS-PAGE and transferred to a nitrocellulose membrane (sc-3718, Santa Cruz Biotechnology). The membrane was blocked with 5% non-fat skim milk powder (Sigma, 70166) in Tris-buffered saline (TBS). The membrane was washed twice in TBST (TBS containing 0.1% Tween-20) and incubated overnight with primary antibody (1 : 500 monoclonal rat anti-caspase-3, sc-7148, anti-b-actin, sc-47778, Santa Cruz Biotechnology). The membrane was then incubated with HRP conjugated secondary antibody (1 : 1000 goat anti-mouse IgG1-HRP, sc-2060, Santa Cruz Biotechnology) for 2 h. The blot was developed with kit reagents (sc-2048, Santa Cruz Biotechnology kit). Data were analyzed using Image J OD analysis software. Signals were normalized with respect to β-actin.
Statistical analysis
Variables were examined using the Kolmogorov-Smirnov test to determine whether they were normally distributed. Data with normal distribution were analyzed using the one-way ANOVA test, whereas data without normal distribution were evaluated using the Kruskal-Wallis test. When overall significance was observed in the ANOVA test, pairwise post-hoc tests were performed using Tukey’s test. The Mann-Whitney U test was performed to assess the significance of pairwise differences after the Kruskal-Wallis test. Scale variables were presented as mean and standard deviation (SD), with 95% confidence intervals (CIs). Statistical analyses were performed using SPSS version 17 (SPSS Inc., Chicago, IL, US). Graph-Pad Prism software, version 6.00 for Windows (Graph-Pad Software, San Diego, CA, US) was used to prepare the figures. An overall 5% type I error level was used to infer statistical significance.
Results
The experimental study was completed without any adverse events and specimens were collected from each animal for histological and biochemical analyses.
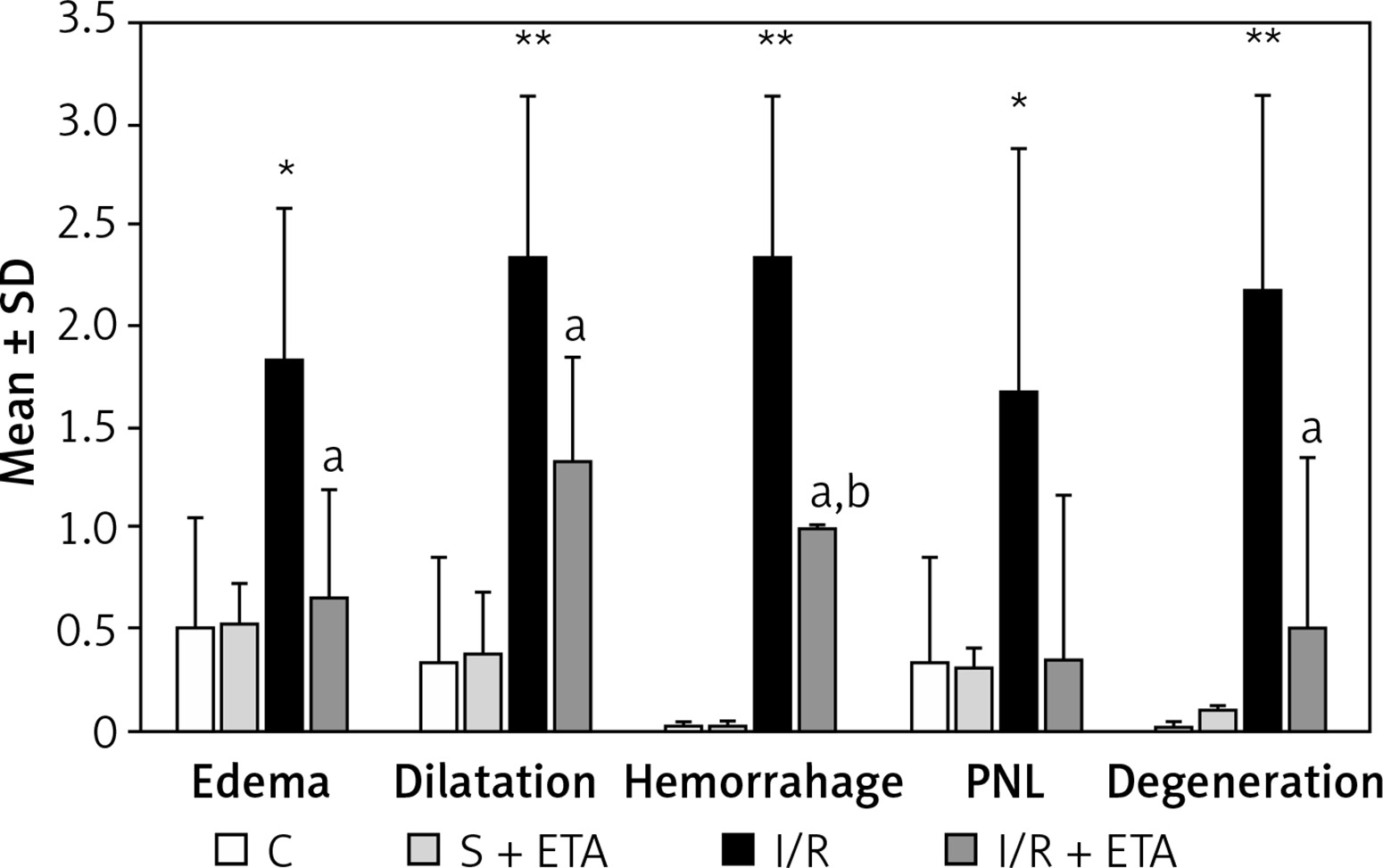
In evaluating histological damage in the I/R group, all parameters were significantly worse than the control group. When we compared the I/R and I/R + etanercept groups, edema, dilatation, hemorrhage, degeneration and PNL infiltration parameters differed significantly (p < 0.05; p < 0.01; p < 0.01, and p < 0.01, p < 0.05, respectively) (Figures 1 A–C, 2).
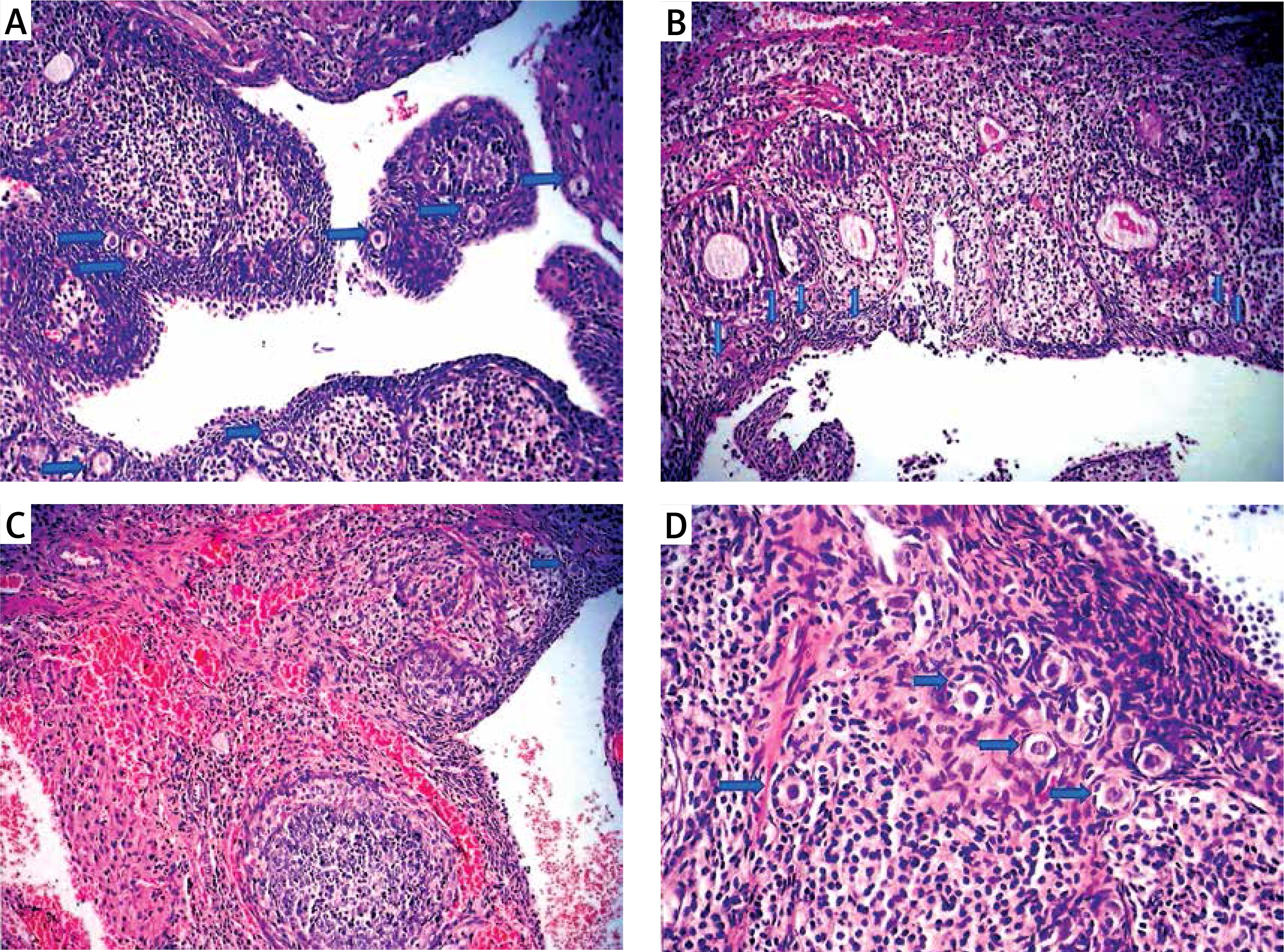
Figure 1
Light microscopic appearance of ovaries (H + E; 100×). A – Normal follicles and corpus luteum were observed in the sham-operated group. B – The I/R group showed vascular dilatation (thick arrow), hemorrhage (thin arrow) follicular degeneration (arrow neck) and polymorphonuclear lymphocyte infiltration (star). C – In the 30 mg/kg ETA group, mild hemorrhage (thin arrow), vascular dilatation (thick arrow), follicular degeneration (arrow neck) and polymorphonuclear lymphocyte infiltration (star) were seen
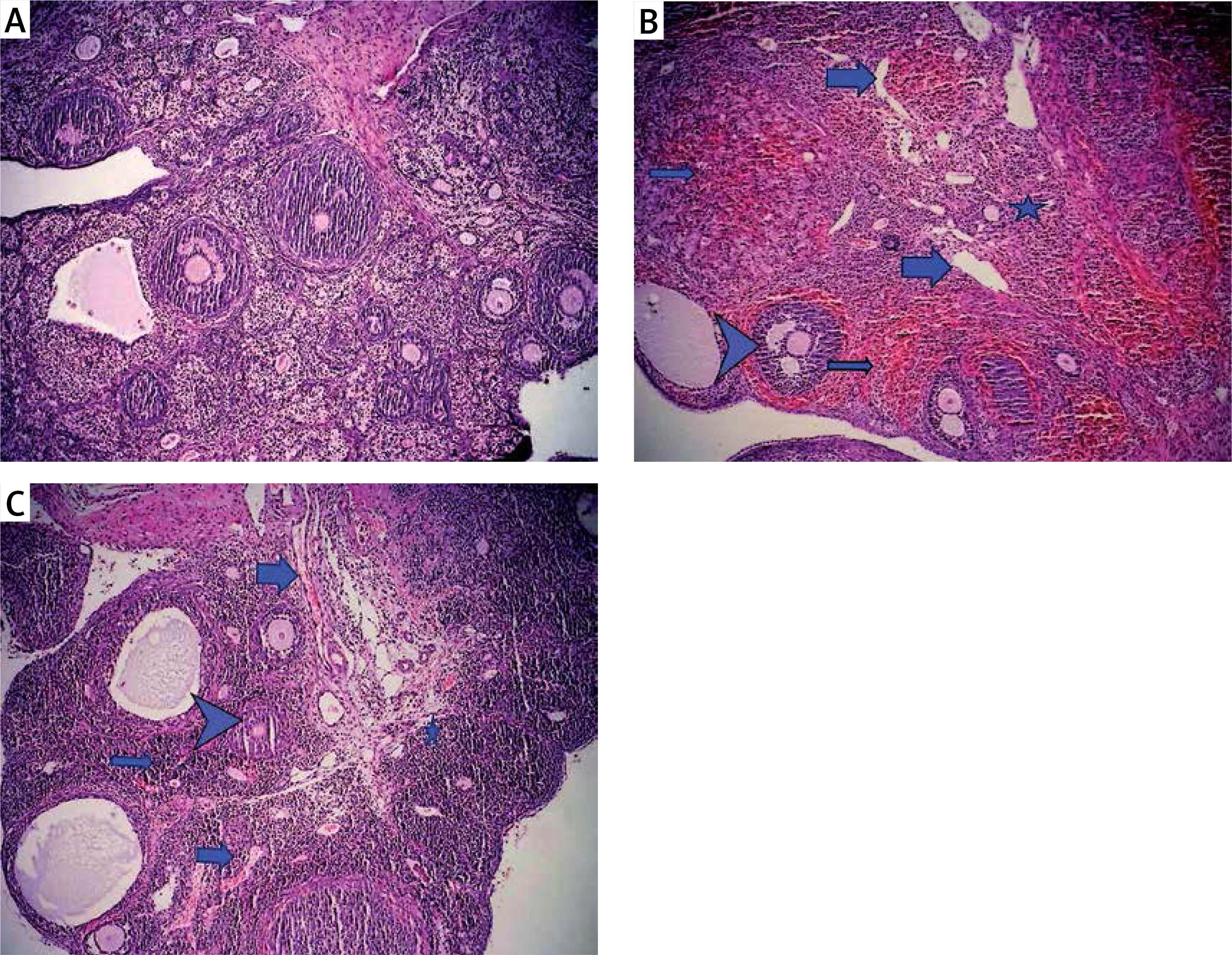
Figure 2
Histologic evaluation scores among study groups
*p < 0.05, **p < 0.01, C vs. I/R; ap < 0.05, I/R + ETA vs. I/R; bp < 0.01, C vs. I/R + ETA.
There were no statistically significant differences in GSH, MDA, MPO, SOD, 8-OHdG, caspase-3 protein activity or AMH levels, as well as follicular counts between sham and sham + etanercept operated groups (sham + etanercept group versus C group, p > 0.05).
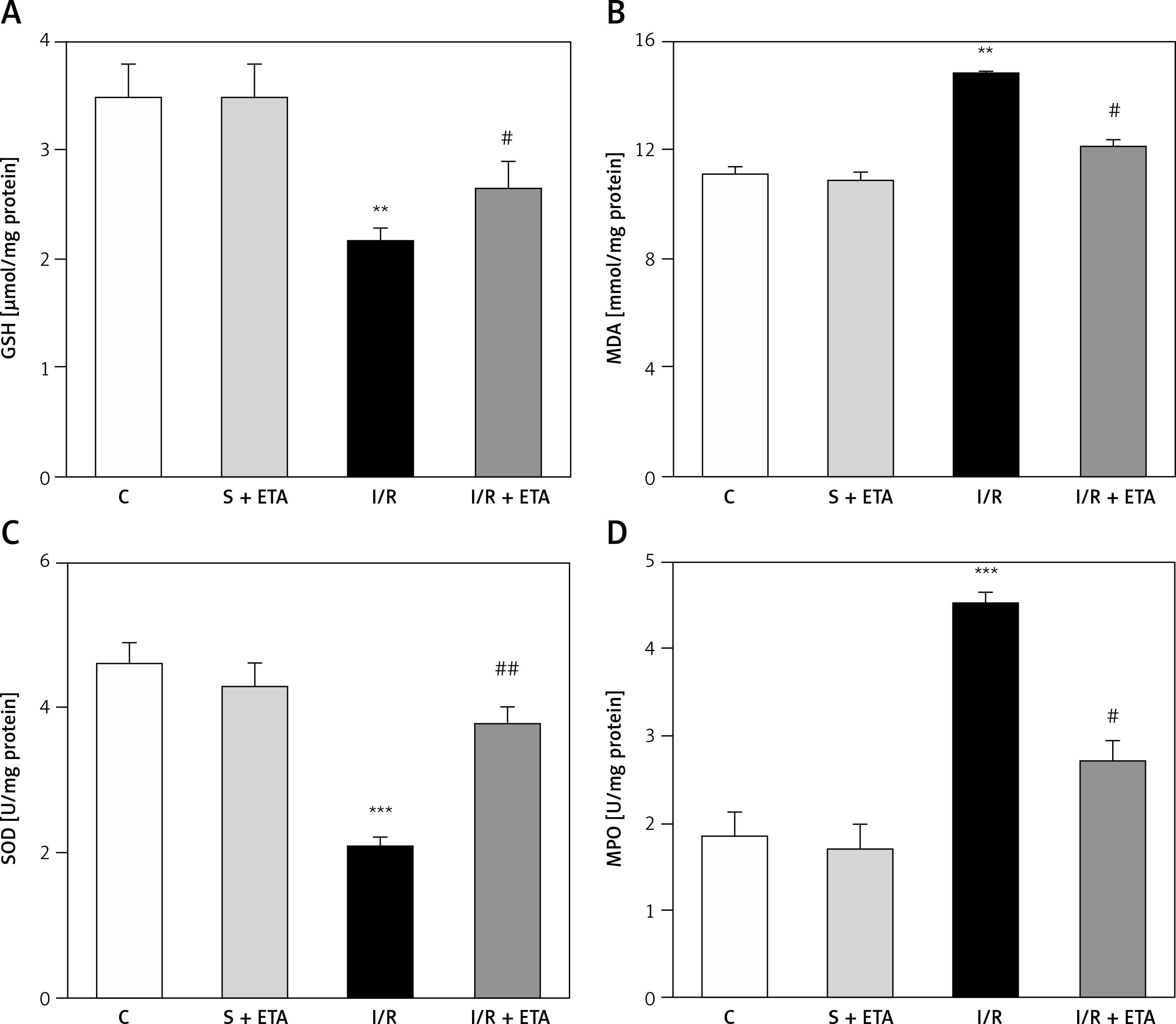
The GSH and SOD levels of the I/R group were compared with the control group and were significantly lower (p < 0.01). The GSH and SOD levels of the I/R + etanercept group were significantly higher than the I/R group (p < 0.05, p < 0.01). The mean MDA and MPO levels of the I/R group were significantly higher than the control group (p < 0.01, p < 0.05), while the MDA and MPO levels of the I/R + etanercept group were significantly lower than the I/R group (p < 0.05) (Figure 3).
Figure 3
Oxidative stress parameters at the tissue level. A – Tissue GSH levels. B – Tissue MDA levels. C – Tissue SOD levels. D – Tissue MPO levels
**p < 0.01, ***p < 0.001 vs. control group, #p < 0.05, ##p < 0.01 vs. I/R group.
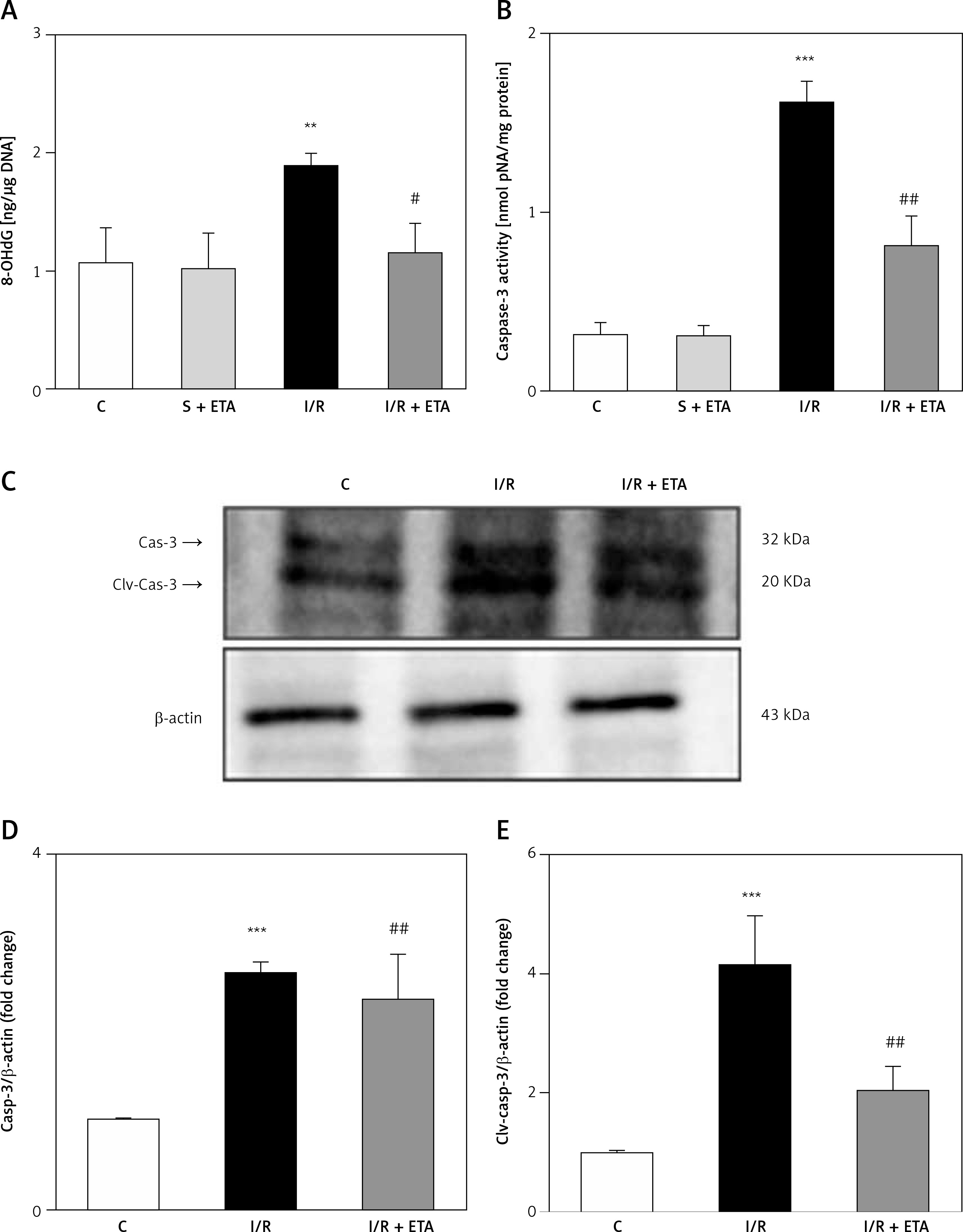
8-OHdG level was significantly higher in the I/R group than the control group (p < 0.01). However, the 8-OHdG level was significantly lower in the I/R + etanercept group than the I/R group (p < 0.05), indicating more severe oxidative DNA damage in the I/R group than the I/R + ETA group (Figure 4). Caspase-3 activity levels of the I/R group were compared with the control group and were significantly higher (p < 0.001). Caspase-3 activity levels in the I/R + etanercept groups were significantly lower than the I/R group (p < 0.01). Caspase-3 protein expression increased after I/R (p < 0.001) and decreased in the I/R + etanercept group (p < 0.01). Cleaved-caspase-3 levels were significantly induced (p < 0.001) by I/R, whereas these values were decreased in the I/R + etanercept group (p < 0.01) (Figure 4).
Figure 4
Apoptotic markers at the tissue level. A – Tissue 8-OHdG levels. B – Tissue caspase-3 activity level. C – Apoptotic caspase protein expression, western blotting band from tissue. D – Densitometry analysis of Cas- 3/B-actin protein expression levels. E – Densitometry analysis of cleaved Cas-3/b-actin protein expression levels compared between groups
**p < 0.001, ***p < 0.01 vs. control group, #p < 0.05,##p < 0.01 vs. I/R group.
Serum AMH levels were significantly decreased when pre-and postoperative values were compared in the I/R and I/R + etanercept groups (p < 0.001, p < 0.001, respectively). The reduction of AMH levels in the I/R + etanercept group was significantly lower than the I/R group (Table I). The mean number of all kinds of follicles was decreased in the I/R group (p < 0.001). No statistically significant difference was observed in the large antral and corpus luteum follicle counts between control and I/R + etanercept groups. The etanercept-treated group of rats had a significantly greater average number of primordial and preantral follicles compared to the I/R group (p > 0.05) (Table I, Figure 5).
Table I
Comparison of follicle counts and pre-/post-operative AMH levels in study groups
| Parameter | Control (6) | I/R (6) | I/R + ETA (6) | P-value |
|---|---|---|---|---|
| Follicle counts: | ||||
| Primordial | 12 ±4.94 | 3.50 ±1.04a | 6.17 ±1.32b | 0.001 |
| Preantral | 6.83 ±2.63 | 2.17 ±0.75a | 5.83 ±0.75b | 0.004 |
| Small antral | 2.83 ±0.75 | 1.67 ±0.51d | 2.67 ±0.51c | 0.017 |
| Large antral | 1.83 ±0.40 | 1.17 ±0.40 | 1.33 ±0.51 | 0.063 |
| Corpus luteum | 4.67 ±1.03 | 2.50 ±0.83a | 3.17 ±0.75c | 0.009 |
| AMH [ng/ml]: | ||||
| Preoperative | 8.74 ±0.78 | 9.34 ±1.03 | 9.11 ±0.66 | 0.480 |
| Postoperative | 8.77 ±0.89 | 4.49 ±0.41e | 7.06 ±0.75b,f | < 0.001 |
| AMH reduction | 0.02 ±0.49 | –4.85 ±0.76e | –2.05 ±0.48e,f | 0.001 |
Discussion
The biochemical and histopathological findings of our study showed that etanercept decreased inflammation related oxidative stress, as well as DNA damage. The significant increase in AMH in the I/R + etanercept group compared to the I/R group demonstrated the protective effect of etanercept on ovarian reserve. This protection may have resulted primarily from inhibition of inflammation and apoptosis.
The inflammatory process is caused by the production of inflammation-related cytokines or chemokines and characterized by leukocyte recruitment to the site of damage. TNF-α and chemokines were observed before a significant leukocyte infiltration in the local inflammation area. The peak levels of TNF-α were already achieved 2 h after challenge. This indicates that local resident cells synthesized the TNF-α, which led to both chemokine synthesis and leukocyte recruitment [16].
In the histologic evaluation of the ovaries, pretreatment with etanercept attenuated the inflammation and leukocyte recruitment by inhibiting TNF- α. Consistent with our research, in myocardial I/R injury Yang et al. found that administration of etanercept inhibited neutrophil infiltration and TNF-α production [17].
Etanercept is commonly used for autoimmune disease and could be considered safe when administered during conception/first trimester [18] and childhood [19]. The anti-oxidant and anti-apoptotic activity of etanercept is well established in rat models [17, 20]. Similarly, we observed a significant decrease in MDA and MPO, which are produced by I/R injury, including a significant increase in GSH and SOD, important antioxidant enzymes in tissues. We suggest that pre-treatment with the TNF-α blocker etanercept attenuated oxidative stress and ovarian injury.
Singlet oxygen and hydroxyl oxygen radicals cause hydroxylation at the C-8 position of 2′-deoxyguanosine to produce 8-OHdG, which is strongly correlated with oxidative DNA damage [21]. We also observed a significant decrease in 8-OHdG levels in the etanercept treatment group. On the other hand, caspase-3 is an important component of caspase cascades that has been associated with apoptosis-dependent signaling pathways. Caspase-3 activation is increased as a result of oxidative stress [20]. Hafez et al. showed that etanercept can have a protective effect with down-regulation of oxidative stress induced caspase-3 expression in hepatotoxicity and nephrotoxicity [22]. It is reported that etanercept reduced TNF-α levels. Therefore pro-apoptotic proteins may be reduced and anti-apoptotic proteins may be induced with treatment [23]. White et al. reported that etanercept treatment can ameliorate proinflammatory and proapoptotic changes in kidney ischemia-reperfusion injury in rats [24]. In agreement with these observations, in our study, etanercept treatment counteracted apoptotic cell death, as measured by immunoblotting for caspase-3 protein expression and caspase-3 activity.
We also evaluated the protective effect of etanercept on ovarian reserve by measuring follicle count and observed that the loss of primordial, preantral, and small antral follicles was smaller in the etanercept group. Apoptosis is one of the most important mechanisms of cell death and therefore etanercept might have provided protection of the follicle pool against IR injury via inhibition of apoptotic pathways.
The AMH is a member of the transforming growth factor superfamily, and is secreted from the granulosa cells of primary growing follicles in the human ovaries until the early antral stage [25]. Serum AMH level is a marker for preantral follicles in the primary pool [26].
In this study, we found AMH levels to be significantly higher in the etanercept-I/R group than the I/R group. It is reported in the literature that the decrease in AMH levels could be responsible for the decrease in the number of growing follicles after reperfusion, through an increase in the production of free oxygen radicals, aggregation of activated neutrophils and cellular membrane and mitochondrial lipid peroxidation [3], which is consistent with our findings.
We were the first to study the effect of etanercept, which possesses antioxidant, anti-apoptotic and ovarian reserve protective properties, on ovarian ischemia reperfusion injury. These data demonstrated that etanercept, through its anti-inflammatory effect, could reverse I/R injury in the ovary.
Some limitations were noted. Like in the literature [27, 28] we administered the medication before induction of I/R injury. On the other hand, in clinical ovarian torsion, we are only able to give medication after ischemia occurs, but our preliminary study may explain the protective effects of etanercept on ovarian reserve and aid in the understanding of how etanercept functions in the I/R process. On the other hand, with this timing of administration of etanercept we had another implication; ovarian tissue may undergo ischemia-reperfusion injury and consequent oxidative damage during ovarian transplantation similar to ovarian torsion, and such agents may be beneficial in the setting of ovarian graft transplantation, as was recently shown by Mahmoodi et al. [29]. Also, for instance, preoperative administration of etanercept may be beneficial in young women who have a high index of suspicion for ovarian torsion.
Another limitation was the absence of data related to long-term alterations in protective effects of etanercept on ovarian tissue.
In conclusion, however, appropriate dose and optimum drug administration period can only be determined with studies on the human reproductive system. Therefore, we recommended that subsequent studies include different doses and drug administration periods in order to define the adequate dose and optimum timing for the prevention of I/R ovarian damage and for protection of reserve.