Introduction
Cardiovascular disease is considered one of the most serious healthy burdens and responsible for a large part of global mortality [1]. The incidence rate of acute myocardial infarction has declined in recent years due to the advanced therapeutic regimens, but the cardiovascular disease-induced mortality has increased mainly because of chronic heart failure (CHF) [2]. Chronic heart failure is an abnormality of cardiac structure or function that results in the heart fails to pump sufficiently to maintain blood flow to meet the body needs [3]. Patients suffering from CHF have some common clinical symptoms, including excessive tiredness, shortness of breath and leg swelling, leading to the decreased quality of life and increased economic burden on health care [4]. It is reported that some risk factors are closely related with the initiation and development of CHF, such as myocardial infarction, myocarditis, high blood pressure, abnormal inflammation, alcohol abuse and myocardial disfunction [5]. Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) belong to the natriuretic peptides and are closely associated with the development of CHF [6]. Although a great progress has been achieved in the management of CHF, the outcomes of the patients remain dismal [7]. Thus, novel therapeutic strategies are urgently needed.
Emodin (1, 3, 8-trihydroxy-6-methylanthraquinoe) is an active constituent of Rheum officinale and Polygonum cuspidatum [8, 9]. Abundant studies indicate that emodin has obviously anti-inflammatory effects in various diseases and abnormal conditions by inhibition of pro-inflammatory cytokines and suppression of T lymphocyte proliferation [10, 11]. Recent research found that emodin serves protective role against some cardiovascular diseases. For example, Song et al. demonstrated that emodin could ameliorate the experimental autoimmune myocarditis in rats [12]. Wu et al. also reported the protective effects of emodin in acute myocardial infarction, which performed by regulation of inflammation [13]. However, the role of emodin in CHF treatment has been rarely investigated.
Emerging studies have focused on the role of microRNAs (miRNAs) in the molecular mechanisms underlying the protective effects of emodin. In lipopolysaccharide-induced injury H9c2 cells, emodin was proved to improve the myocarditis by regulating miR-223 and the downstream inflammatory cytokines [14]. In myocardial infarction cell model, emodin could ameliorate the hypoxia-induced injury through upregulation of miR-138 [15]. A significant low expression of miR-26b-5p has been reported in the study by Vegter et al. in the patients with CHF compared with healthy individuals [16]. In addition, miR-26b has also been found to improve myocardial infarction by relieving inflammatory response in mice [17]. However, the biological function of miR-26b in the development of CHF and its relationship with emodin remain unclear.
In this study, we sought to investigate the protective role of emodin and explore the underlying mechanisms by examining its regulatory effects on miR-26b-5p expression and inflammation in the rats with CHF.
Material and methods
Construction of rat model
The protocols for the animal experiments were approved by the local committee of The Second People’s Hospital of Lianyungang, and all the rats were treated following the Guide for the Care and Use of Laboratory Animals of the Institute for Laboratory Animal Research. The study was conducted in accordance with the Basic & Clinical Pharmacology & Toxicology policy for experimental and clinical studies [18]. A total of 64 10-weeks-old Wistar rats with body weight of 250 ±30 g were obtained from Shanghai Laboratory Animal Center (Shanghai, China). All the rats were housed in a standard environment with free access to water and food and a 12 h light/dark cycle at 25oC. The rats were randomly grouped into 8 groups with the grouping details listed in Table I. Chronic heart failure animal model was constructed by coronary artery ligation as previously described. The rats were randomly grouped into two groups: sham group and CHF model group, and were anesthetized with 50 mg/kg of sodium pentobarbital. In the CHF group, a 6-0 suture was used to ligate the left coronary artery near its branch point from aorta, between the left atrium and the pulmonary artery outflow tract. In the sham group, the rats were treated the same as the CHF group without the ligation of their coronary arteries. After the surgery, all the rats received penicillin for 3 days to avoid infection. To regulate the expression of miR-26b-5p, the rats in the CHF group received caudal vein injection with miR-26b-5p mimic, miR-26b-5p inhibitor, mimic negative control (mimic NC) or inhibitor negative control (inhibitor NC) as the previously described [19]. All the vectors were synthesized by GenePharma (Shanghai, China).
Table I
Treatment for different animal groups
Emodin treatment
The CHF model rats were divided into emodin group and normal saline (NS) group. The rats in the emodin group received 50 mg/kg emodin (ref: E7881, purity ≥ 90%; Sigma, Aldrich, USA) every day for 3 weeks by intragastric administration, while the rats in the NS group were treated with NS using the same dosage.
Determination of cardiac function
To measure the cardiac function of rats, the major indicators obtained from echocardiography and hemodynamic test were estimated. The rats were anesthetized using sodium pentobarbital under the electrocardiograph monitoring. The echocardiography was conducted using a PST 64A sector scanner (8-MHz probe), and the ejection fraction (EF) and fractional shortening (FS) were calculated. For the hemodynamic test, a catheter filled with heparin was inserted into the left ventricle of the rats to connect the pressure transducer and the carrier amplifier. The left ventricular systolic pressure (LVSP) and left ventricular end diastolic pressure (LVEDP) were recorded, and the maximum of the first differentiation of left ventricular pressure (+LV dP/dtmax) was calculated.
Enzyme-linked immune sorbent assay (ELISA)
The expression levels of ANP and BNP and pro-inflammatory cytokines, including IL-1β, IL-6 and TNF-α, in the serum of the rats were measured by ELISA. The concentration of proteins was evaluated by a BCA assay kit (Thermo Fisher Scientific, Waltham, MA, USA), and the protein levels of ANP, BNP, IL-6 and TNF-α were examined using an ELISA kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturers’ instruction. The absorbance at 450 nm was measured to calculate the final protein levels.
Cell culture and transfection
Rat cardiomyocyte cell line H9c2 was purchased from the Cell Bank of the Chinese Academy of Science (Shanghai, China). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) added with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Waltham, MA, USA) at 37oC in a humidified atmosphere with 5% CO2. For cell transfection, miR-26b-5p mimic or mimic NC were transfected into the cells by Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturers’ instruction. The cells after 48 h of transfection were used for the verification of target gene of miR-26b-5p.
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the heart tissues by TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and examined using a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA) regarding the purity and concentration of the RNA. An ABI miRNA reverse transcription kit (Applied Biosystem, Foster City, USA) was used to synthesize cDNA from the RNA. To analyze the expression value of miR-26b-5p, qPCR was performed using a Power SYBR green kit (Thermo Fisher Scientific, Waltham, MA, USA) and an ABI 7500 Real-Time PCR System (Applied Biosystem, Foster City, USA). U6 was used as an endogenous control gene. The final relative expression of miR-26b-5p was calculated by 2–ΔΔCt method and normalized to U6.
Luciferase reporter assay
This study preliminarily analyzed the target gene of miR-26b-5p. Phosphatase and tensin homolog (PTEN) was predicted as a potential target gene of miR-26b-5p using TargetScan (http://www.targetscan.org/vert_72/). Thus, luciferase reporter assay was carried out to confirm the target gene. The wild-type (WT) 3’-untranslated region (3’-UTR) or mutant type (MT) 3’-UTR of PTEN were integrated into the firefly luciferase reporter vectors with Rnilla luciferase (Promega, Fitchburg, WI, USA). The H9c2 cells were seeded into 24 well plates and co-transfected with the recombinant vector and miR-26b-5p mimic, miR-26b-5p inhibitor, mimic NC or inhibitor NC. The luciferase activity was detected after 48 h of transfection using a SecrePair Dual-Luciferase Reporter System (Promega).
Statistical analysis
SPSS statistical software (SPSS Inc., Chicago, IL) and GraphPad Prism software (GraphPad Software, Inc, USA) were used to perform all the statistical analyses, and the data were expressed as mean ± SD. Student’s t test and one-way ANOVA were used to compare the data compliance with normal distribution between groups. A p-value of less than 0.05 was considered statistically significant.
Results
Emodin improves the cardiac function of the chronic heart failure rats
In this study, the CHF rat model was constructed by coronary artery ligation. The cardiac function of the rats was evaluated using the indicators shown in Table II. The results revealed that the levels of ANP and BNP and LVEDP were increased, while the EF, FS, LVSP and +LV dP/dtmax were decreased in the CHF rats compared with the sham group (all p < 0.05), indicating that the CHF model was successfully obtained. After the administration of emodin, the decreased EF, FS, LVSP and +LV dP/dtmax and the increased ANP and BNP and LVEDP induced by CHF were all rescued by the treatment of emodin (all p < 0.05).
Table II
Effects of emodin on cardiac function of chronic heart failure rats
| Parameters | Sham (n = 8) | CHF (n = 8) | NS (n = 8) | Emodin (n = 8) |
|---|---|---|---|---|
| ANP [ng/l] | 38.2 ±5.5 | 77.6 ±4.9** | 78.8 ±4.7** | 43.2 ±4.1## |
| BNP [ng/l] | 66.4 ±5.1 | 129.2 ±10.2** | 131.8 ±9.3** | 71.6 ±6.2## |
| EF (%) | 80.4 ±7.1 | 61.4 ±5.4* | 59.6 ±4.0* | 77.8 ±6.0# |
| FS (%) | 52.2 ±5.0 | 30.8 ±4.8* | 28.8 ±5.3* | 48.8 ±5.3# |
| LVSP [mm Hg] | 104.4 ±20.8 | 64.8 ±10.4* | 62.6 ±9.5* | 85.7 ±10.5# |
| LVEDP [mm Hg] | 2.3 ±1.1 | 21.6 ±4.8** | 20.7 ±4.9** | 5.5 ±2.4## |
| +LV dp/dtmax [mm Hg/s] | 5466.0 ±292.7 | 3248.4 ±281.2* | 3128.2 ±274.6* | 4565.4 ±360.2# |
CHF – chronic heart failure, ANP – atrial natriuretic peptide, BNP – brain natriuretic peptide, EF – ejection fraction, FS – fractional shortening, LVSP – left ventricular systolic pressure, LVEDP – left ventricular systolic pressure, +LV dP/dtmax – maximum of the first differentiation of left ventricular pressure.
Emodin suppresses the inflammation of the chronic heart failure rats
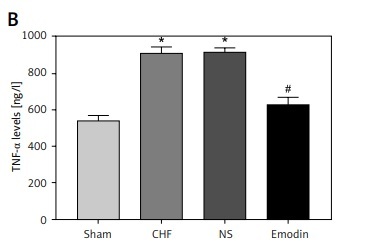
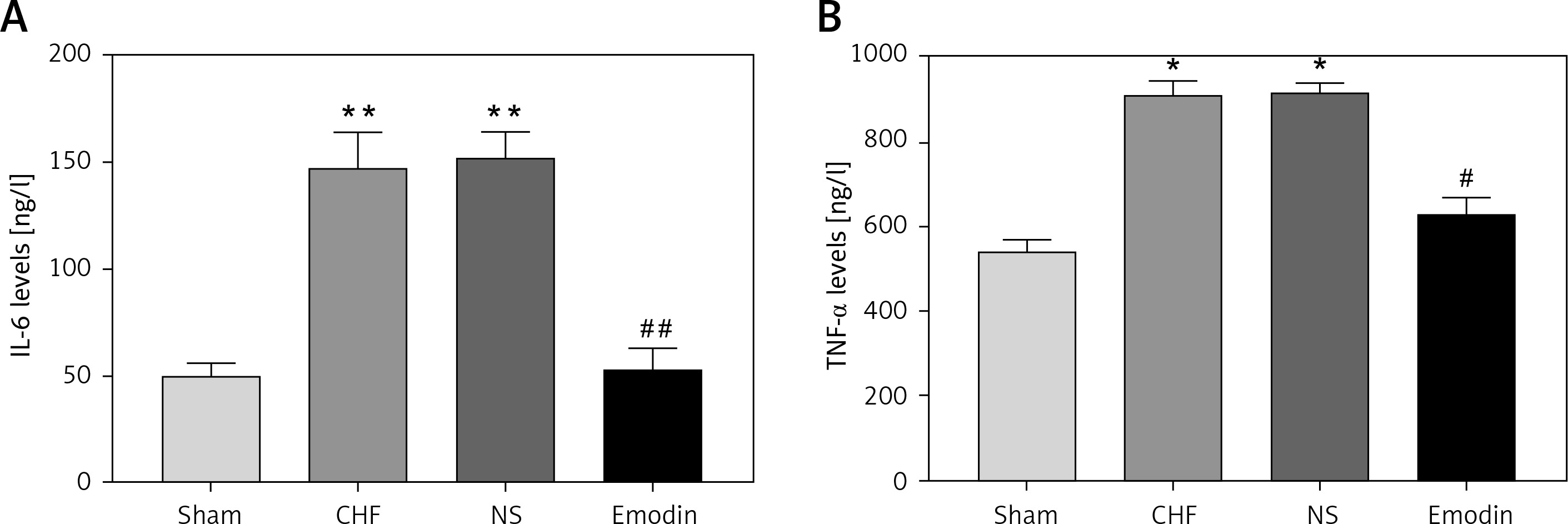
Considering the pivotal role of inflammation in the development of CHF, the serum concentrations of IL-6 and TNF-α were measured. As expected, the levels of IL-6 and TNF-α were both significantly upregulated after the CHF modeling (all p < 0.05). Notably, the increases in IL-6 and TNF-α levels by CHF were abrogated by the treatment of emodin (all p < 0.05, Figure 1).
Figure 1
Proinflammatory cytokine levels in chronic heart failure (CHF) rats before and after treatment with emodin. A – Emodin could reduce the CHF-induced increased IL-6 concentration. B – The elevated TNF-α level induced by CHF was decreased by emodin. Each group included 8 animals. *P < 0.05, **p < 0.01 vs. sham group; #p < 0.05, ##p < 0.01 vs. CHF group
Expression of miR-26b-5p in the chronic heart failure rats before and after emodin treatment
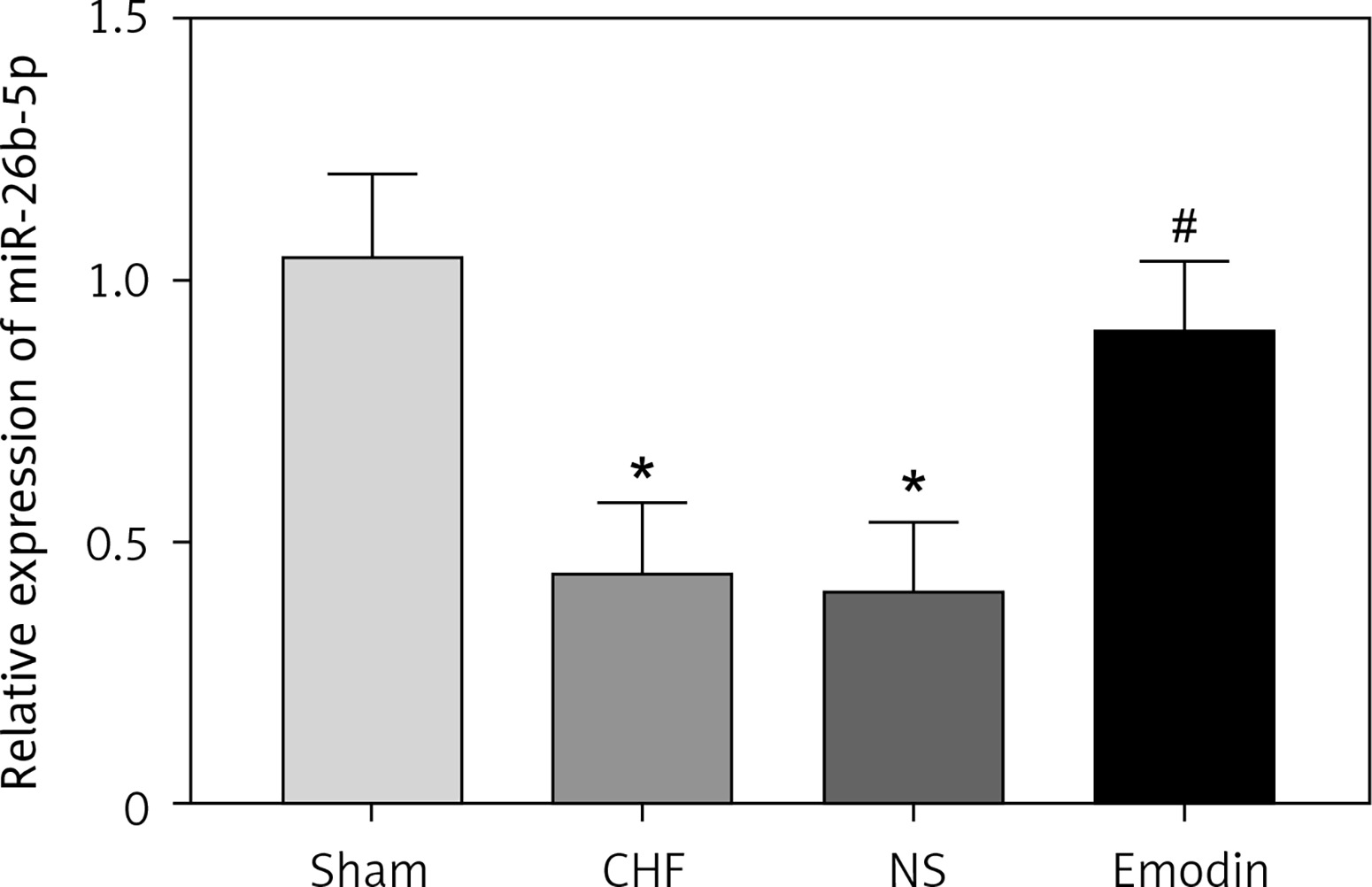
It is reported that the expression of miR-26b-5p was decreased in CHF. In consistent with the previous result, we also found the downregulated expression of miR-26b-5p in the CHF rats compared with the sham group (p < 0.05). Interestingly, we observed that the decreased expression of miR-26b-5p in the CHF model was remarkably elevated by the treatment of emodin (p < 0.05, Figure 2).
Figure 2
Expression of miR-26b-5p in chronic heart failure (CHF) rats before and after emodin administration. Expression of miR-26b-5p was upregulated in CHF, but was suppressed after treatment with emodin. Each group included 8 animals. *P < 0.05 vs. sham group; #p < 0.05 vs. CHF group
Overexpression of miR-26b-5p improves the cardiac function and inflammation of the chronic heart failure rats
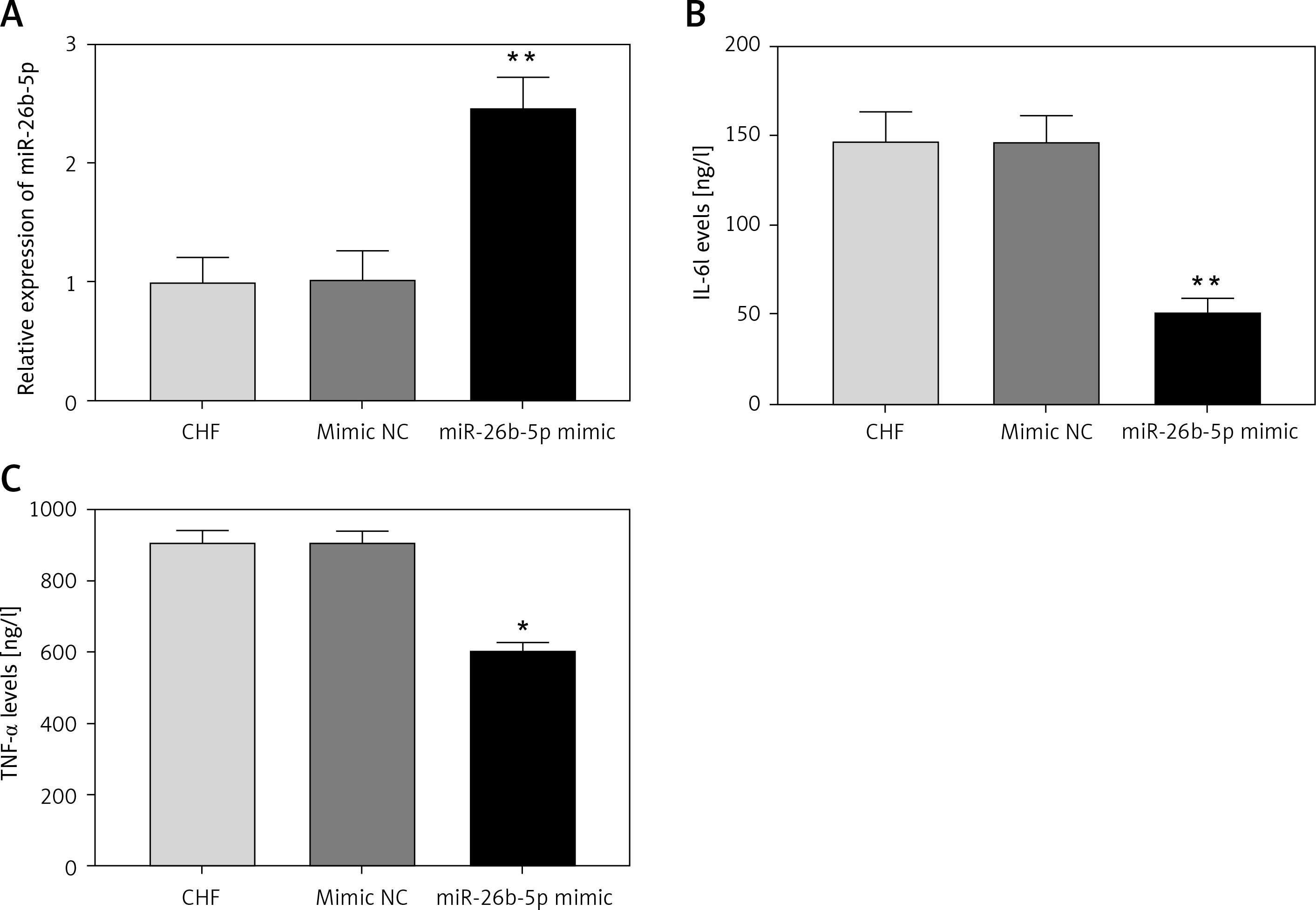
Given the decreased expression of miR-26b-5p in CHF rats, we examined its effects on cardiac function and inflammation. The expression of miR-26b-5p was upregulated by the animal transduction with miR-26b-5p (p < 0.01, Figure 3 A). As shown in Table III, EF, FS, LVSP and +LV dP/dtmax in the CHF model were promoted by the overexpression of miR-26b-5p, while ANP and BNP levels and LVEDP were suppressed by the upregulation of miR-26b-5p (all p < 0.05). For the examination of inflammatory response, we found that the overexpression of miR-26b-5p in the CHF rats resulted in significantly reduced IL-6 and TNF-α concentrations (all p < 0.05, Figures 3 B, C).
Table III
Effects of miR-26b-5p on the cardiac function in chronic heart failure rats
| Parameters | CHF (n = 8) | Mimic NC (n = 8) | miR-26b-5p mimic (n = 8) |
|---|---|---|---|
| ANP [ng/l] | 77.6 ±4.9** | 75.6 ±5.7 | 47.2 ±3.4** |
| BNP [ng/l] | 129.2 ±10.2** | 130.6 ±9.1 | 69.0 ±5.6** |
| EF (%) | 61.4 ±5.4* | 59.0 ±5.1 | 75.0 ±5.4* |
| FS (%) | 30.8 ±4.8* | 29.0 ±5.1 | 48.4 ±5.2* |
| LVSP [mm Hg] | 64.8 ±10.4* | 64.8 ±12.4 | 92.3 ±18.5* |
| LVEDP [mm Hg] | 21.6 ±4.8** | 21.5 ±5.4 | 5.9 ±2.3** |
| +LV dP/dtmax [mm Hg/s] | 3248.4 ±281.2* | 3156.4 ±262.5 | 4827.2 ±338.4* |
CHF – chronic heart failure, ANP – atrial natriuretic peptide, BNP – brain natriuretic peptide, EF – ejection fraction, FS – fractional shortening, LVSP – left ventricular systolic pressure, LVEDP – left ventricular systolic pressure, +LV dP/dtmax – maximum of the first differentiation of left ventricular pressure.
Figure 3
Effects of miR-26b-5p on inflammation in chronic heart failure (CHF) rats. A – Expression of miR-26b-5p was upregulated by miR-26b-5p mimic. B, C – Upregulation of miR-26b-5p in CHF rats resulted in decreased IL-6 and TNF-α levels. Each group included 8 animals. *P < 0.05, **p < 0.01 vs. CHF group
MiR-26b-5p is involved in the protective effects of emodin in the chronic heart failure rats
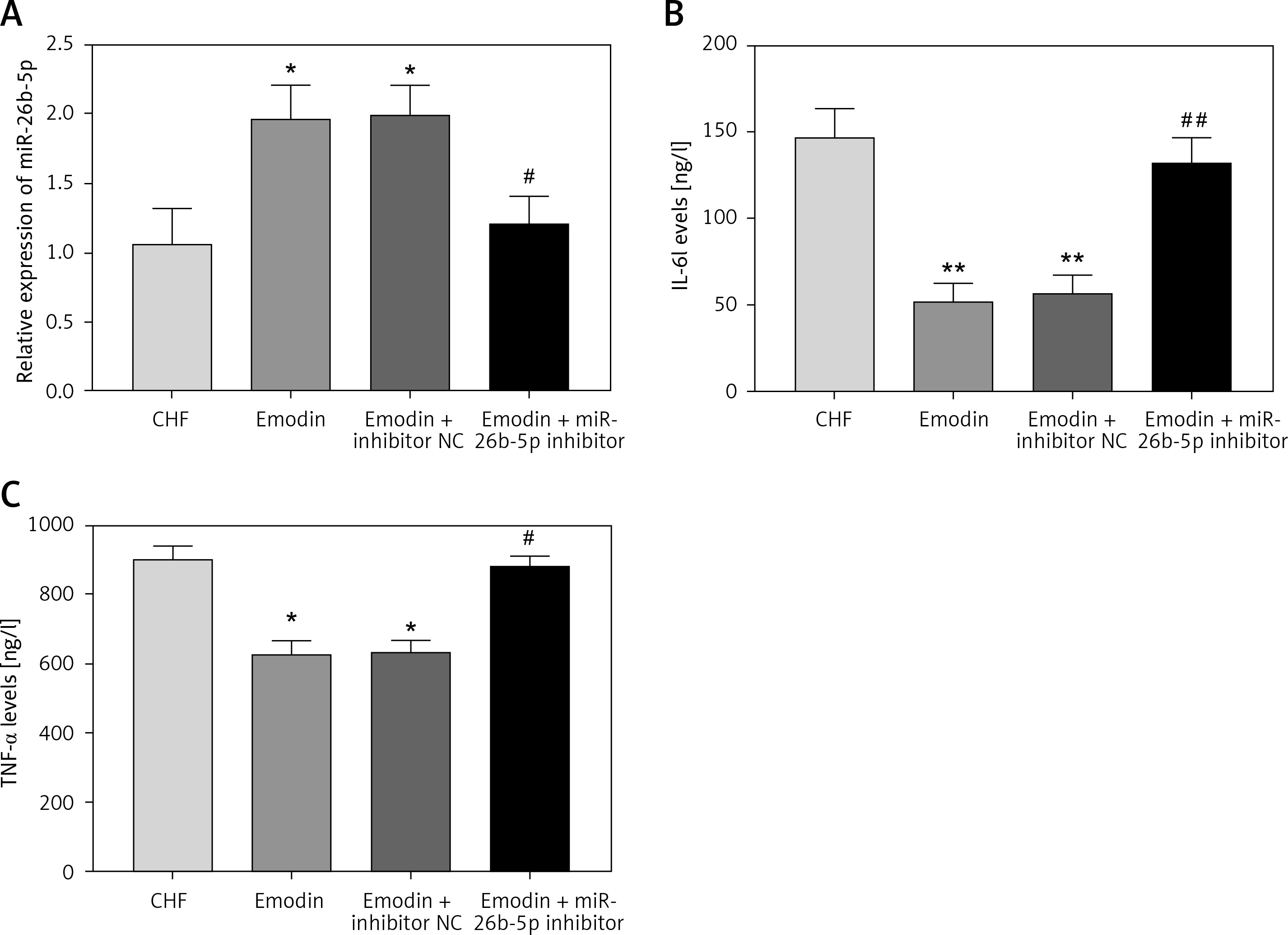
Since the regulatory effect of emodin on the expression of miR-26b-5p, we suspected that miR-26b-5p might be involved in the protective effects of emodin against CHF. Thus, the miR-26b-5p inhibitor was adopted to suppress the expression of miR-26b-5p in the CHF rats treated with emodin. The results in Figure 4 A revealed that the increased expression of miR-26b-5p induced by emodin was successfully reduced by miR-26b-5p inhibitor (p < 0.05). From Table IV, the emodin-induced increased EF, FS, LVSP and +LV dP/dtmax and the decreased ANP, BNP levels and LVEDP were all abrogated by the knockdown of miR-26b-5p (all p < 0.05). In addition, the reduction of miR-26b-5p could enhance the emodin-induced downregulation in IL-6 and TNF-α levels (all p < 0.05, Figures 4 B, C).
Table IV
miR-26b-5p acted as a mediator in the protective effects of emodin on cardiac function in chronic heart failure rats
| Parameters | CHF (n = 8) | Emodin (n = 8) | Emodin + inhibitor NC (n = 8) | miR-26b-5p inhibitor (n = 8) |
|---|---|---|---|---|
| ANP [ng/l] | 77.6 ±4.9 | 43.2 ±4.1** | 43.0 ±5.3** | 74.4 ±4.8# |
| BNP [ng/l] | 129.2 ±10.2 | 71.6 ±6.2** | 71.4 ±5.9** | 120.2 ±7.2## |
| EF (%) | 61.4 ±5.4 | 77.8 ±6.0* | 76.8 ±4.1* | 63.8 ±5.4# |
| FS (%) | 30.8 ±4.8 | 48.8 ±5.3* | 48.6 ±5.2* | 36.0 ±4.7# |
| LVSP [mm Hg] | 64.8 ±10.4 | 85.7 ±10.5* | 85.7 ±10.9* | 69.1 ±11.7# |
| LVEDP [mm Hg] | 21.6 ±4.8 | 5.5 ±2.4** | 6.1 ±2.8** | 18.5 ±4.2## |
| +LV dP/dtmax [mm Hg/s] | 3248.4 ±281.2 | 4565.4 ±360.2* | 4632.0 ±413.2* | 3461.0 ±313.6# |
CHF – chronic heart failure, ANP – atrial natriuretic peptide, BNP – brain natriuretic peptide, EF – ejection fraction, FS – fractional shortening, LVSP – left ventricular systolic pressure, LVEDP – left ventricular systolic pressure, +LV dP/dtmax – maximum of the first differentiation of left ventricular pressure.
Figure 4
Inhibition of miR-26b-5p rescued the effects of emodin on inflammation in chronic heart failure (CHF) rats. A – Expression of miR-26b-5p was downregulated by miR-26b-5p inhibitor. B, C – The reduced levels of IL-6 and TNF-α induced by emodin were elevated by knockdown of miR-26b-5p. Each group included 8 animals. *P < 0.05, **p < 0.01 vs. CHF group; #p < 0.05, ##p < 0.01 vs. Emodin group
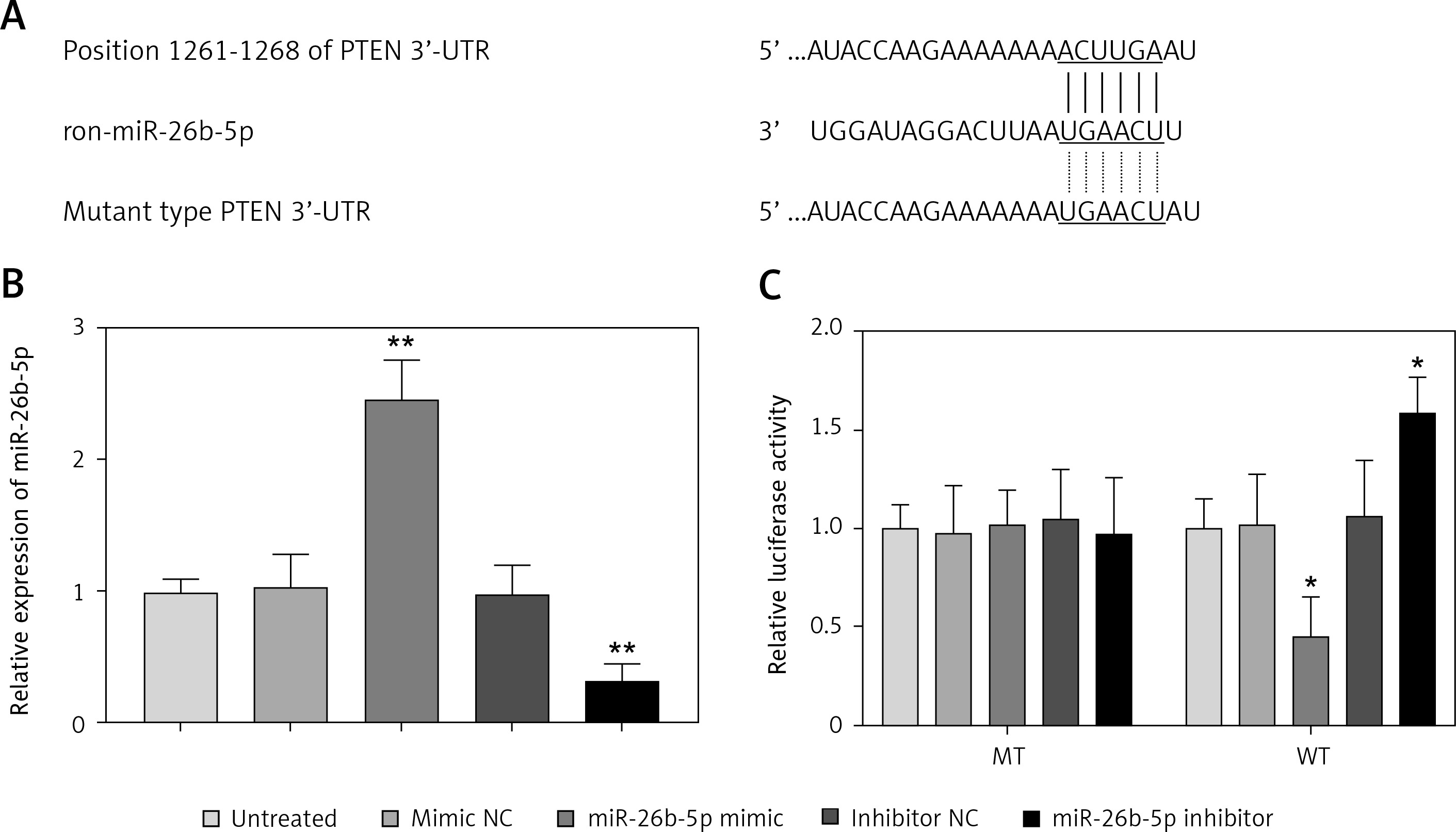
Phosphatase and tensin homolog acts as a target for miR-26b-5p in cardiomyocytes
In the 3’-UTR of PTEN, we found the complementary sequences of miR-26b-5p (Figure 5 A). Thus, we used luciferase reporter assay to confirm the interaction between miR-26b-5p and PTEN in the rat cardiomyocyte cell line H9c2. As shown in Figure 5 B, the expression of miR-26b-5p in the cells was upregulated by the miR-26b-5p mimic, but was downregulated by the miR-26b-5p inhibitor (all p < 0.01). The luciferase activity results in the WT 3’-UTR of PTEN group were significantly inhibited by the overexpression of miR-26b-5p, whereas were promoted by the knockdown of miR-26b-5p (all p < 0.05, Figure 5 C). However, no effect of miR-26b-5p was observed on the luciferase activity in the MT group.
Figure 5
PTEN served as a target gene of miR-26b-3p in H9c2 cells. A – A complementary sequence of miR-26b-5p was predicted in the 3ʹ-UTR of PTEN. B – Expression of miR-26b-5p was upregulated by miR-26b-5p mimic, but was downregulated by miR-26b-5p inhibitor in H9c2 cells. C – Overexpression of miR-26b-5p suppressed the luciferase activity, but the downregulation of miR-26b-5p promoted the luciferase activity in the WT group. *P < 0.05, *p < 0.01 vs. untreated group
Discussion
Traditional Chinese medicine has attracted increasing attention in the treatment of cardiovascular diseases, especially in atherosclerotic cardiovascular disease and CHF [20, 21]. For example, Xian et al. performed a multicenter randomized double-blind placebo-controlled trial to investigate the effect of yangxinkang tablets on CHF, and found that yangxinkang tablets could improve CHF regarding the cardiac function and quality of life in CHF patients [22]. Another study by Li et al. also prove the clinical efficacy of qili qiangxin capsules in the patients with CHF [23]. As an effective constituent of Rheum officinale, emodin has been investigated in some human disease, especially in cardiovascular diseases. In atherosclerosis, emodin was demonstrated to suppress the inflammatory response and acted an anti-atherosclerotic role [24]. In myocardial infarction, emodin could ameliorate this disease by inhibiting the inflammation and cell apoptosis [13]. In myocarditis, emodin was also involved in the regulation of proinflammatory cytokines IL-1β and TNF-α, leading to inhibited inflammation in the rats with myocarditis, and thereby improved this disease [12]. However, rarely report regarding the role of emodin in the development of CHF. In the present study, we constructed the CHF model using coronary artery ligation in rats, and found that the cardiac function was impaired after the modeling. After the treatment of emodin, the impaired cardiac function was improved, which was indicated by the increased EF, FS, LVSP and +LV dP/dtmax and the decreased ANP and BNP levels and LVEDP. Thus, we considered that emodin had significantly protective effects against CHF in rats.
It is known that inflammation plays critical role during the development of various diseases [25–28], especially in cardiovascular diseases, such as myocardial infarction [29], myocarditis [30] and also CHF [31]. Emodin has been determined as an important regulator of inflammation. It could suppress the inflammation in various diseases, such as jejunum injury [32] and acute lung injury and acute respiratory distress syndrome [33]. In myocardial infarction and myocarditis, emodin exerts its protective effects also through the regulation of inflammation [12, 13]. In the present study, we focused on the influences of emodin on the concentration of proinflammatory cytokines IL-6 and TNF-α, and found that the increased inflammation induced by CHF was obviously suppressed by the treatment of emodin. It is implied that emodin might improve CHF by regulation of inflammation.
MiRNAs are a group of small noncoding RNAs and play crucial roles in various diseases, including cardiovascular diseases [34]. The clinical application of aberrant expression of key molecules especially for miRNAs has attracted increasing attention in disease treatment [35, 36]. In recent years, the role of miRNAs acted in the mechanisms of the protective effects of emodin has been demonstrated. For instance, emodin was demonstrated to attenuate myocarditis via downregulation of miR-223 [14]. MiR-138 was proved to mediate the protective effects of emodin against myocardial infarction [15]. In pancreatic cancer, emodin was found to inhibit the cancer cell EMT and invasion by upregulating the expression of miR-1271 [37]. In our study, we observed a markedly decrease in the expression of miR-26b-5p in CHF rats compared with the controls. More importantly, the reduced miR-26b-5p in the CHF model was promoted by the treatment of emodin. Additionally, the cardiac function of the CHF rats was remarkably improved by the upregulation of miR-26b-5p, and the improved cardiac function induced by emodin was abrogated by the downregulation of miR-26b-5p, which implied that miR-26b-5p might be involved in the development of CHF and participate the effects of emodin.
Previous studies have reported that miR-26b-5p was implicated in inflammation [38]. In our study, the inflammatory response was improved by the overexpression of miR-26b-5p evidenced by the reduced IL-6 and TNF-α concentration. Furthermore, we found that the reduced levels of inflammatory cytokines attributed to emodin treatment were rescued by the knockdown of miR-26b-5p, indicating that emodin improved the inflammation of CHF rats through upregulating miR-26b-5p. To further explore the molecular mechanisms of emodin in CHF treatment, the target gene of miR-26b-5p was predicated and PTEN as a potential target was demonstrated in this study. PTEN is an upstream regulator of the PI3K/AKT signaling, and this signaling has been reported to mediate the protective effects of Qishenkeli against myocardial apoptosis in CHF [39, 40]. Thus, we suspected that the emodin might improve the cardiac function and inflammation by regulation of miR-26b-5p and the downstream PTEN/PI3K/AKT signaling pathway in CHF rats. This hypothesis needs to be verified in further studies.
In conclusion, all the data in this study revealed that emodin plays a protective role against the impaired cardiac function and dysregulation in inflammation in CHF rats, and its effects may be achieved by regulating the miR-26b-5p/PTEN pathway. Our discovery indicates that emodin has an obviously potential as an anti-CHF therapeutic agent.