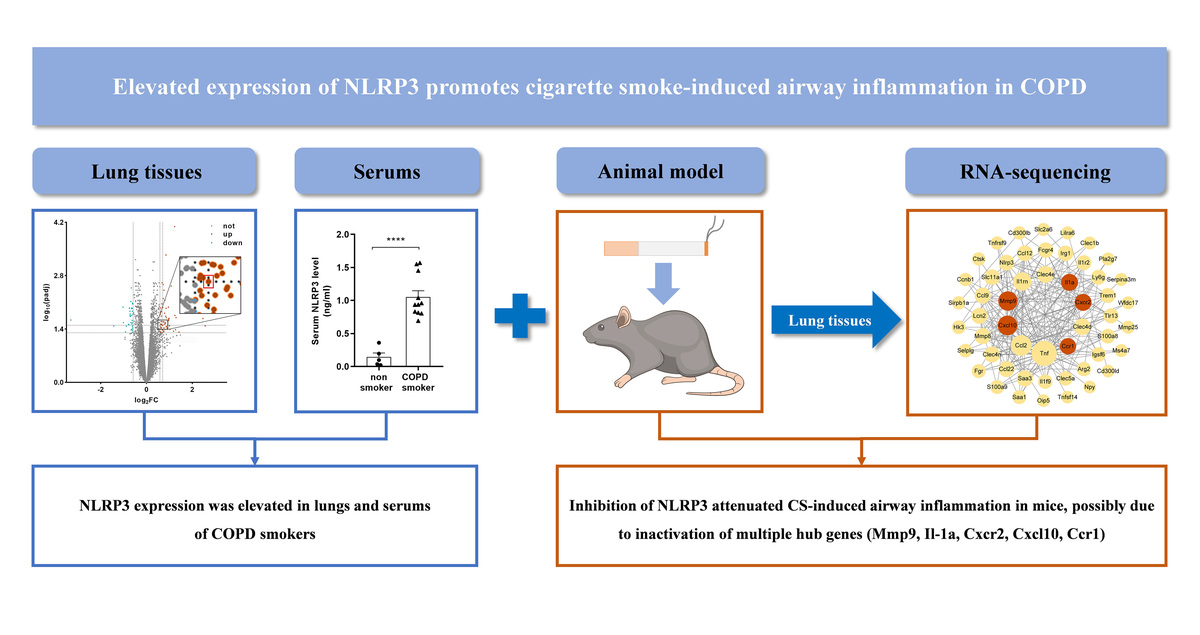
Introduction
Chronic obstructive pulmonary disease (COPD), the third leading cause of disease-related death worldwide, is characterized by progressive airflow limitation, owing to chronic airway inflammation [1]. To relieve the inflammatory level in airways, inhaled corticosteroid combined with bronchodilators has been recommended for decades, but pulmonary function decline in COPD could not be improved, especially in end-stage patients [2]. Lung transplantation seems to be the optimal solution for advanced COPD, but a shortage of donor organs and immunological complications limit its clinical application. Noticeably, it has been reported that stem cell therapy and tissue engineering may be promising substitutions for lung transplantation, which shed new light on non-drug treatment for advanced COPD [3, 4]. However, it should be emphasized that early diagnosis based on spirometry with interventions regarding airway inflammation in COPD, particularly induced by cigarette smoke (CS), is vital for COPD management [5].
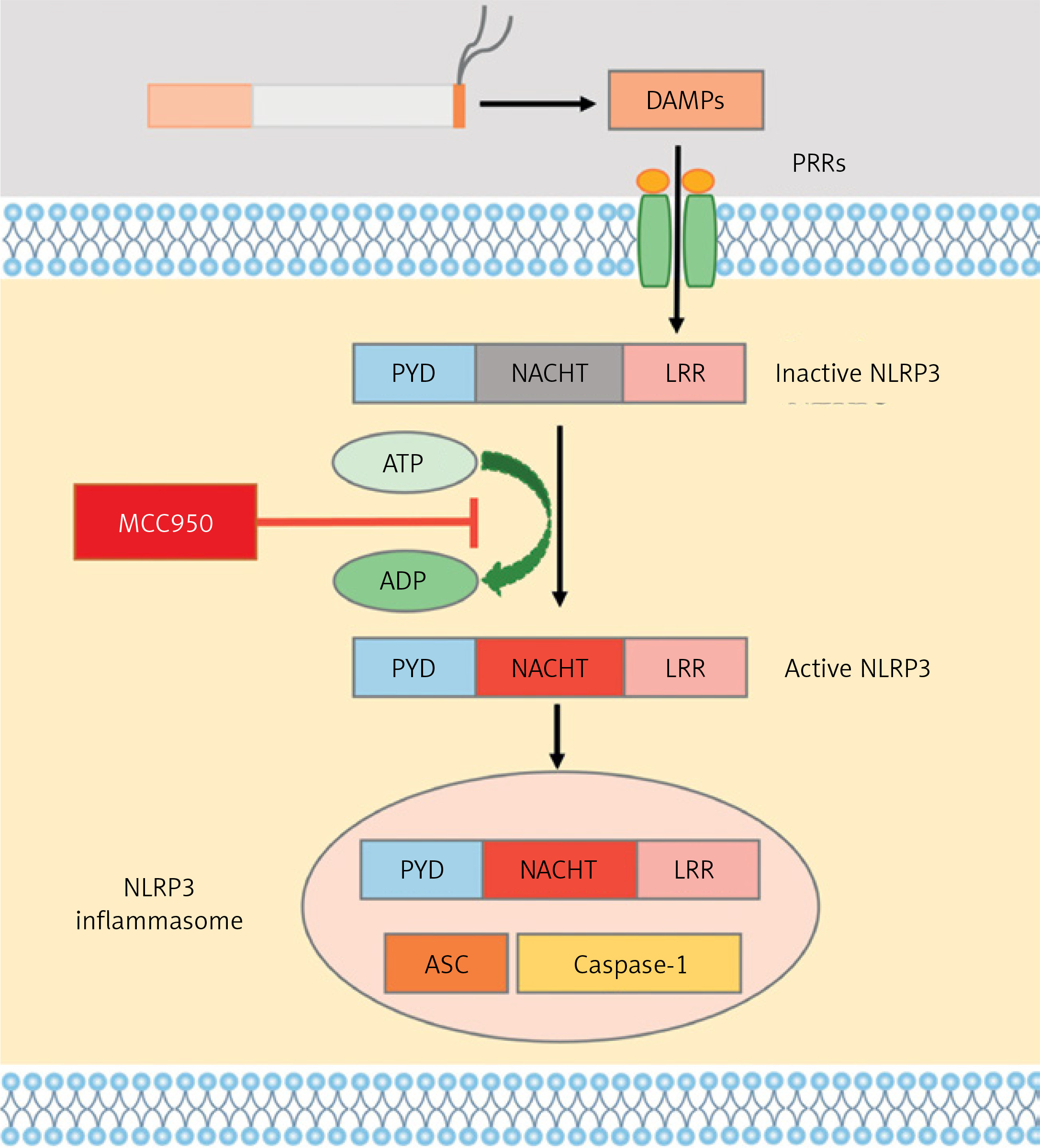
NOD-like receptor protein 3 (NLRP3), a functional protein encoded by the gene located at chromosome 1q44 [6], is composed of three domains: the amino-terminal pyrin domain (PYD), the central nucleotide binding domain (NACHT) and carboxy-terminal leucine-rich repeats (LRR) [7]. As a member of pattern recognition receptors (PRRs), NLRP3 can be activated by damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs) or other stimuli, which results in either NLRP3-dependent pyroptosis or an inflammatory response in the host by forming the NLRP3 inflammasome, containing activated NLRP3, ASC (apoptosis-associated speck-like protein containing CARD) and protease caspase-1 (Figure 1) [8, 9].
Figure 1
Diagram of formation of NLRP3 inflammasome. PAMPs stimulated by CS exposure, via binding pattern recognition receptors (PRRs) on targeted cell membrane, activated NLRP3, followed by formation of NLRP3 inflammasome, which can be blocked by MCC950, a specific inhibitor of NLRP3 on ATP-to-ADP transformation
The relationship between NLRP3 activation and COPD pathogenesis has been discussed recently. NLRP3 and other components in the NLRP3 inflammasome (caspase-1 and ASC), and NLRP3-dependent proinflammatory cytokines (interleukin (IL)-18 and IL-1β) were all significantly up-regulated in stable COPD patients, especially those with acute exacerbation [10, 11]. Moreover, CS exposure could remarkably increase NLRP3 expression, as well as acute/subacute airway inflammation in mice, which was reversed by NLRP3 gene knockout or pharmacological inhibition of NLRP3 [12–14]. Overall, these data indicated NLRP3 could play a key role in the CS-induced airway inflammation in COPD. However, the underlying mechanisms in this process still need more illustration.
Herein, in this study, we initially determined NLRP3 expression in lung tissues and sera from COPD smokers and non-smokers. To further explore the role of NLRP3 in CS-induced airway inflammation in COPD, a CS-exposed mouse model of airway inflammation was established, through treatment with MCC950, a specific inhibitor of NLRP3. MCC950 can block the ATP hydrolysis motif of the NLRP3 NACHT domain and leave the complex in an inactive form, thereby inhibiting the downstream response and function (Figure 1) [15, 16]. Subsequently, RNA sequencing with bioinformatic analyses was performed to reveal the underlying mechanisms.
Material and methods
Microarray data and analysis
The GSE37768 (18 COPD smokers and 9 healthy non-smokers) and GSE103174 (37 COPD smokers and 10 healthy non-smokers) datasets regarding gene expression data of COPD smokers and non-smokers were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The GSE37768 and GSE103174 datasets were derived from the GPL570 platform of Affymetrix Human Genome U133 Plus 2.0 Array and the GPL13667 platform of Affymetrix Human Genome U219 Array, respectively. All samples from the two different datasets were obtained from human lung tissues.
These datasets were backgrounded corrected and normalized using the R “limma” package and converted to gene symbols according to the probe annotation files in the dataset. A merged dataset composed of GSE37768 and GSE103174 datasets, including 55 COPD smokers and 19 non-smokers, was then generated for further analysis [17]. Batch variability between platforms was eliminated using the combat function of the R “SVA” package [18]. Gene expression data were analyzed for differentially expressed genes (DEGs) using the R “limma” package. DEGs were selected based on the thresholds of q (adjusted p-value) < 0.05 and |log2 FC| > 0.585 (FC > 1.5 or FC < 0.67).
Enzyme-linked immunosorbent assay (ELISA) in serum
Sera were obtained from a case-control cohort, including 13 COPD smokers and 6 healthy non-smokers (Table I). The diagnosis of COPD was based on the criteria of the Global Initiative for Chronic Obstructive Lung Disease [2]. NLRP3 level in sera was determined according to the instructions of a commercial ELISA kit (Jianglai Biological Technology Co. Ltd., Shanghai, China). This study was approved by the Biomedical Ethics Committee of West China Hospital of Sichuan University (approval number: 2023289) and the written informed consent was obtained from the participants.
Table I
Clinical data of subjects
| Parameter | Non-smokers | COPD smokers |
|---|---|---|
| Number | 6 | 13 |
| Age [years] | 48 (32–63) | 62.84 (52–74) |
| Gender (M/F) | 4/2 | 13/0 |
| Smoking [pack-years] | No | 29.59 (0.5–49) |
| FEV1 [l]) | 2.97 (1.21–5.48) | 1.66 (0.71–2.66)* |
| FVC [l] | 3.87 (1.72–6.76) | 3.17 (1.99–4.36) |
| FEV1% predicted | 96.29 (58.33–124.2) | 59.08 (24.2–93.4)** |
| FEV1/FVC% | 75.79 (70.37–81.15) | 51.53 (25.63–67.61)*** |
Animal model and treatment
Wild-type C57BL/6J male mice 6–8 weeks old (Beijing HFK Bioscience Co., Ltd., China) were randomly divided into five groups, with 5 mice per group, as follows: saline (SA) group, CS group, MCC950 (10 mg/kg) group, CS + MCC950 (5 mg/kg) group and CS+MCC950 (10 mg/kg) group. They were specific pathogen-free and were bred in a temperature-controlled (22°C) room with a 12 : 12 h light-dark cycle. All mice had free access to food and water. Animal experiments involved in this study strictly followed the regulations of the Animal Ethics Committee of West China Hospital of Sichuan University (approval number: 20230211009).
After a week acclimatization period, mice in the CS and CS + MCC950 groups were exposed to Marlboro cigarettes (Marlboro, Philips Morris, USA), with 1.1 mg of nicotine and 11 mg of tar per cigarette, in a custom-designed nose-only exposure tube, with a concentration of particulate matter of 320–340 mg/m3 [19]. Briefly, the restrained mice were fixed to a chamber, in which the nose of the mouse was exposed to the smoke for 75 min twice a day, 5 days per week for 4 weeks, while the SA and MCC950 groups were exposed to filtered air [20]. Mice in the MCC950 and CS + MCC950 groups were intraperitoneally injected with 5 mg/kg or 10 mg/kg of MCC950 three times per week (Monday, Wednesday and Friday), 30 min before CS exposure [21], and the SA and CS groups were injected with the same volume of saline according to the same schedule.
Cell counting and ELISA in bronchoalveolar lavage (BAL) fluid
Mice were sacrificed by exsanguination from the right ventricle on the day after the last exposure. The right lungs were lavaged with 0.5 ml of sterile ice-cold PBS supplemented with protease inhibitors (MCE) three times, and the recovery rate was 80% at least. The lavage was centrifuged, and the supernatant was prepared for ELISA, while the cell pellet was resuspended in 500 µl of ice-cold PBS, then the total cell counts in BAL fluid were determined by a hemocytometer (ORFLO). Differential cell counts (neutrophils, macrophages and lymphocytes) were performed on the cell smears stained by the Wright-Giemsa approach. Five different random views of each slice (at least 200 cells in each view) were recorded by a microscope (Nikon, Japan). Two blinded, independent and experienced investigators (MY and JC) took part in the differential cell counts [22]. The NLRP3-dependent proinflammatory cytokines in the supernatant, including IL-1β and IL-18, were measured by ELISA, according to the instructions of commercial ELISA kits (Neobioscience Technology Co. Ltd., Shenzhen, China).
Histologic inflammatory score
The left lung, not lavaged, was fixed in 4% paraformaldehyde (pH 7.4) and then embedded in paraffin, sectioned (4 µm), and stained with hematoxylin and eosin (HE) solution for further histological scoring. Briefly, the perivascular (0–5 points), peribronchial (0–5 points), and parenchymal (0–5 points) infiltration as well as epithelial damage (0–5 points) in each slide was assessed and scored independently by two experienced investigators (JC and YS) according to the previous report [22].
Transcriptomic studies
The right lungs from mice (n = 3 per group) were randomly selected for RNA sequencing. Total RNA was extracted using TRIzol reagent (Invitrogen, CA, USA). After purification and qualification by the NanoDrop 2000 spectrophotometer (Thermo Scientific, USA), the RNA integrity was assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The libraries were constructed using the VAHTS Universal V6 RNA-seq Library Prep Kit, and sequenced on the Illumina Novaseq 6000 platform (Oebiotech Co. Ltd., Shanghai, China). After removal of low-quality reads from raw reads by fastp, HISAT2 was used to map clean reads to the reference genome, calculate fragments per kilobase of exon per million fragments mapped (FPKM), and obtain read counts according to HTSeq-count [20]. Subsequently, PCA analysis and hierarchical cluster analysis of differentially expressed genes (DEGs) were performed by R software package (v3.2.0) to evaluate biological duplication of samples and the expression pattern of genes in different groups.
Bioinformatic analyses
Genes with an absolute log2 fold change > 1 and adjusted p-value < 0.05 were defined as DEGs. The up-regulated genes (CS vs. SA) and down-regulated genes (CS + MCC950 10 mg/kg vs. CS) were intersected and visualized using a Venn diagram by R software package (v3.2.0). To further identify the hub genes among the intersected DEGs, protein-protein interact (PPI) network and Molecular Complex Detection (MCODE, one of the plug-ins in Cytoscape) were then performed and visualized with String (https://cn.string-db.org/) and Cytoscape (v3.9) [23]. Finally, GO and KEGG enrichment analyses (https://cloud.oebiotech.cn/task/) regarding the hub genes were performed.
Real-time quantitative polymerase chain reaction (qPCR)
Real-time qPCR was performed to validate the results of the bioinformatic analyses. Total RNA was extracted from the lavaged lung tissues (right lungs) with the miRNeasy Mini Kit (Qiagen, Germany) following the manufacturer’s instructions. Next, cDNA was prepared with a reverse transcription kit (Takara, Japan). mRNA expression of the hub genes as well as the reference gene (glyceraldehyde-3-phosphate dehydrogenase, GAPDH) was quantified on a LightCycler 96 SW 1.1 instrument (Roche, Switzerland) using a real-time qPCR assay (Roche). The thermal cycle conditions were as follows: a 10-minute pre-incubation period at 95°C, followed by 38 cycles of a two-step amplification (10 s at 95°C, 15 s at 60°C). The primer sequences used in this study are listed in Supplementary Table SI. All data were normalized to GAPDH gene expression, and the relative expression level of each gene was indicated by 2–ΔΔCq.
Statistical analysis
The data were presented as mean ± standard error (SEM) or median with interquartile range. The unpaired t-test was used to compare two groups; otherwise one-way analysis of variance followed by Tukey’s multiple comparisons test was adopted. Statistical analysis was performed with GraphPad Prism version 8.0 (GraphPad Software, La Jolla, CA, USA). Significance was defined by a p-value of 0.05.
Results
NLRP3 was elevated in the lung tissues and sera of COPD smokers
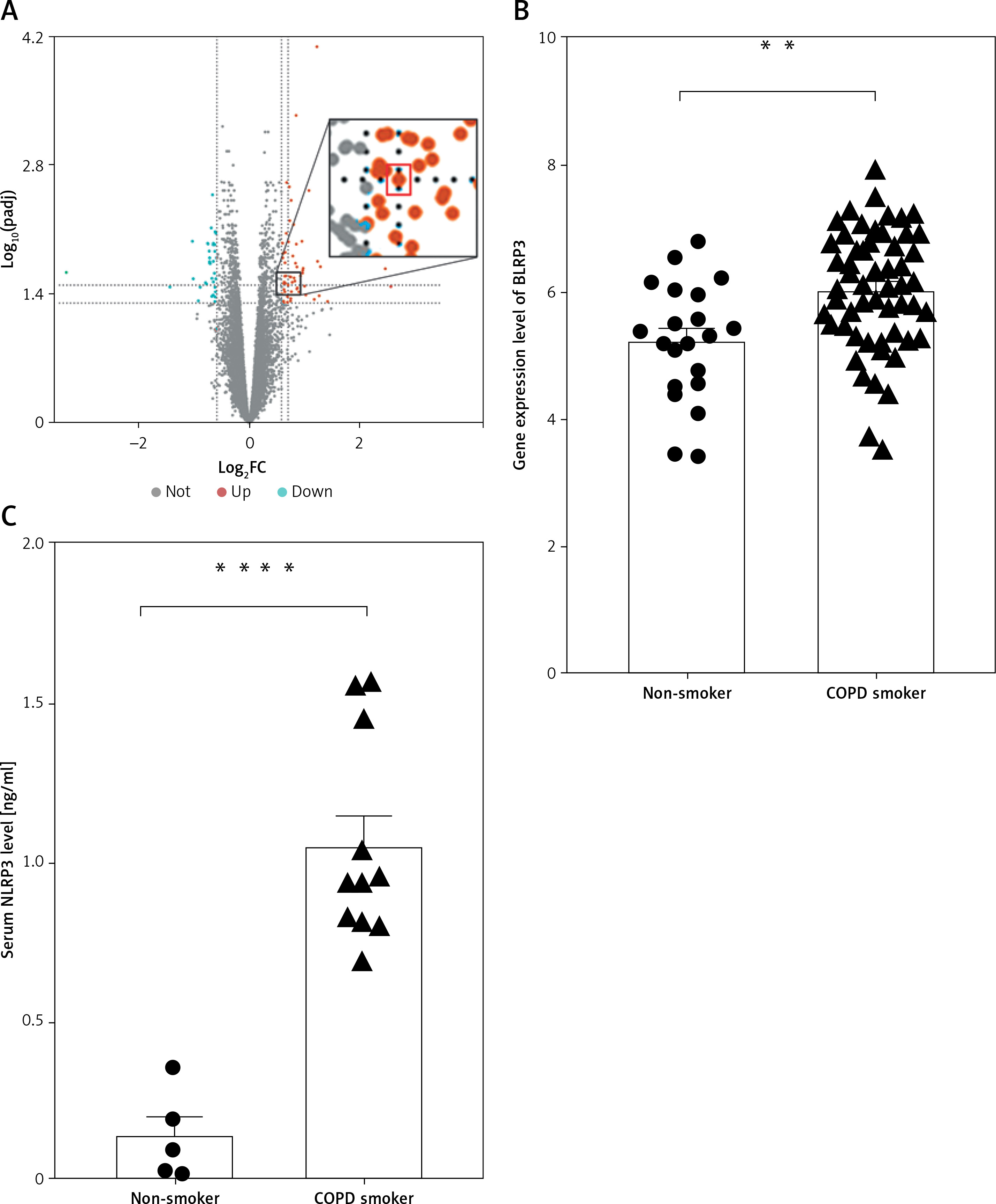
Based on the GEO datasets, a total of 92 DEGs were identified between COPD smokers and non-smokers, among which up-regulated NLRP3 was detected (Figure 2 A). Then, NLRP3 gene expression was further analyzed with the expression matrix, which indicated that NLRP3 was significantly elevated in lung tissues of COPD smokers compared with non-smokers (Figure 2 B). Validation of NLRP3 levels in sera from a case-control cohort confirmed that NLRP3 was remarkably higher in COPD smokers than that in non-smokers (Figure 2 C, Table I).
Figure 2
NLRP3 was significantly elevated in lung tissues and sera of COPD smokers. A – Volcano plot showing the log2 fold change (X-axes) plotted against log10-adjusted p-values (Y-axes) for COPD smokers and non-smokers, with up- and down-regulated DEGs being highlighted in red and green respectively. The spot in the red box within the enlarged image represented up-regulated NLRP3. B – NLRP3 gene expression between COPD smokers and non-smokers based on the expression matrix in the GEO datasets. C – NLRP3 level in sera between non-smokers and COPD smokers by ELISA. Data are presented as mean ± SEM. The significance of differences was examined by unpaired t-test. P < 0.05 was considered statistically significant. *p < 0.05, **p < 0.01, ***p < 0.001, ****p ≤ 0.0001
Inhibition of NLRP3 ameliorated CS-induced airway inflammation in mice
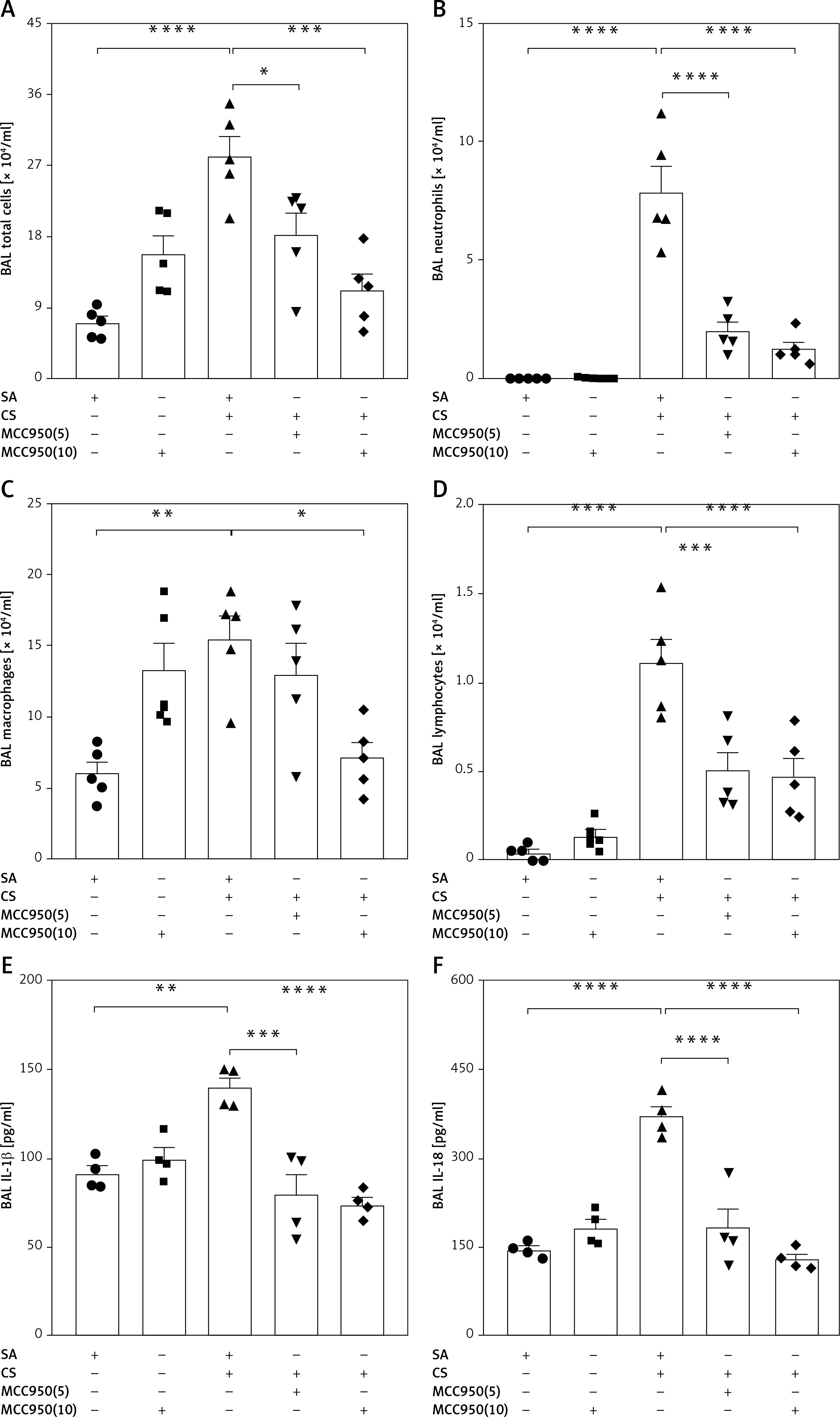
To explore the role of NLRP3 in CS-induced airway inflammation, mice were exposed to CS or air for 4 consecutive weeks with SA or MCC950 treatment. In contrast to the SA group (Figure 3 A), CS-exposed mice showed profound inflammatory cell infiltration around the vessels, bronchia and parenchyma, as well as epithelial damage (Figure 3 C), which was accompanied with higher histological inflammatory scores in HE staining (Figure 3 F), and a significant increase in total (Figure 4 A) and differential (Figures 4 B–D) cell counts and proinflammatory cytokine level (Figures 4 E, F) in BAL fluid. However, MCC950 dose-dependently attenuated CS-induced airway inflammation (Figures 3 D, E), and enhancements in histological inflammatory scores (Figure 3 F), as well as total cell (Figure 4 A), macrophage (Figure 4 B), neutrophil (Figure 4 C), lymphocyte (Figure 4 D) counts, and IL-1β (Figure 4 E) and IL-18 levels (Figure 4 F) in BAL fluid.
Figure 3
MCC950 ameliorated CS-induced airway inflammation in mice. Representative HE-stained images from SA group (A), MCC950 (10) group (B), CS group (C), CS + MCC950 (5) group (D) and CS+MCC950 (10) group (E) are displayed. Scale bars = 100 μm. F – MCC950 treatment attenuated HE histological scores induced by CS exposure. The Arabic numbers in brackets represent the doses of MCC950. Data are presented as mean ± SEM, and the significant differences were examined by one-way analysis of variance followed by Tukey’s multiple comparisons test. P < 0.05 was considered statistically significant. *P < 0.05, **p < 0.01, ***p < 0.001, ****p ≤ 0.0001
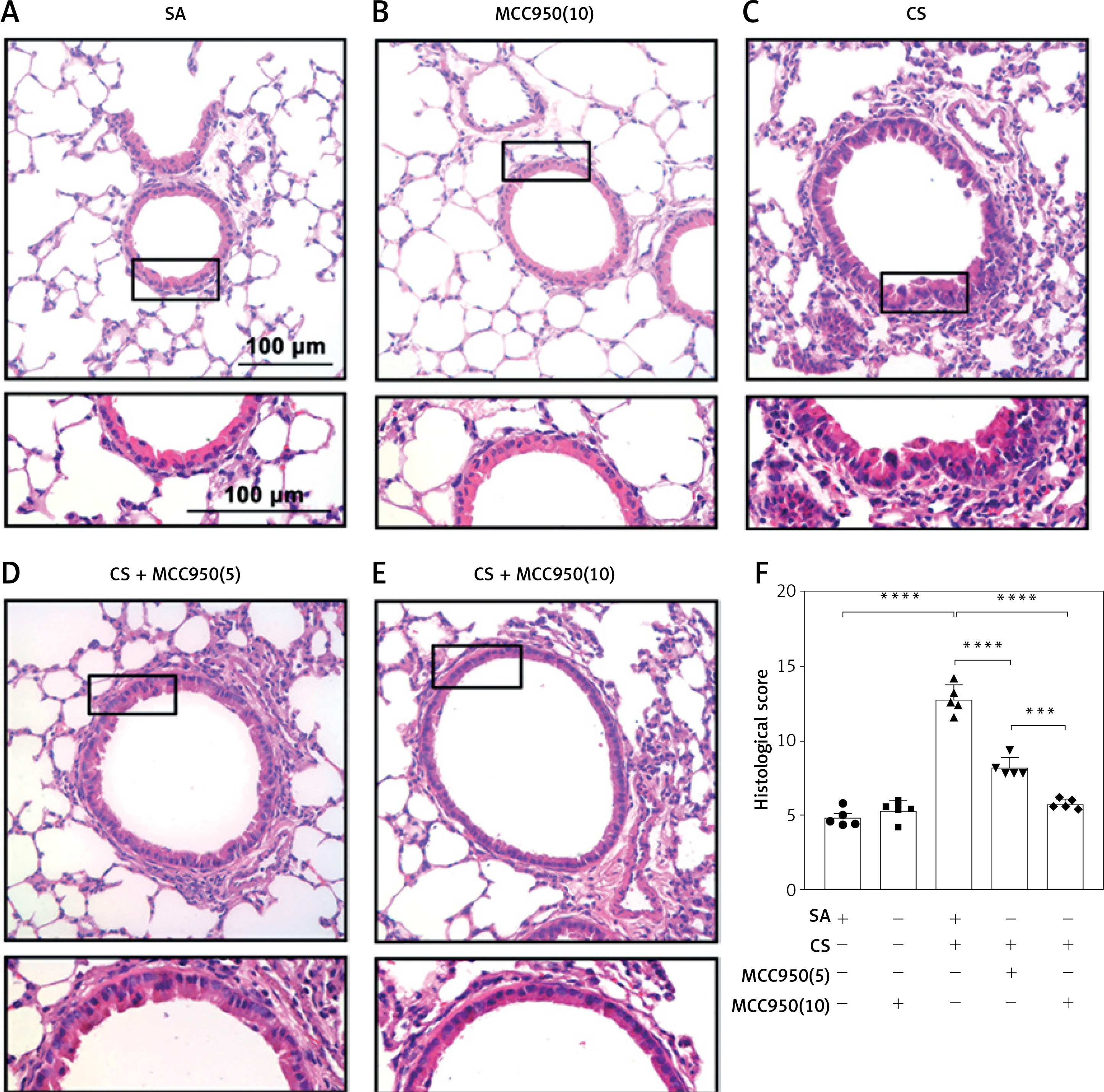
Figure 4
MCC950 ameliorated CS-induced inflammatory cell counts and NLRP3-dependent proinflammatory cytokines in BAL fluid. 4-week CS exposure enhanced the counts of total cells (A), neutrophils (B), macrophages (C), lymphocytes (D), as well as IL-1β (E) and IL-18 (F) in the BAL fluid, which were all attenuated by MCC950 treatment dose-dependently. Data are presented as mean ±SEM. The significance of differences was examined by one-way analysis of variance followed by Tukey’s multiple comparisons test. P < 0.05 was considered statistically significant. *P < 0.05, **p < 0.01, ***p < 0.001, ****p ≤ 0.0001
Mechanisms correlated with NLRP3 in CS-induced airway inflammation in bioinformatic analyses
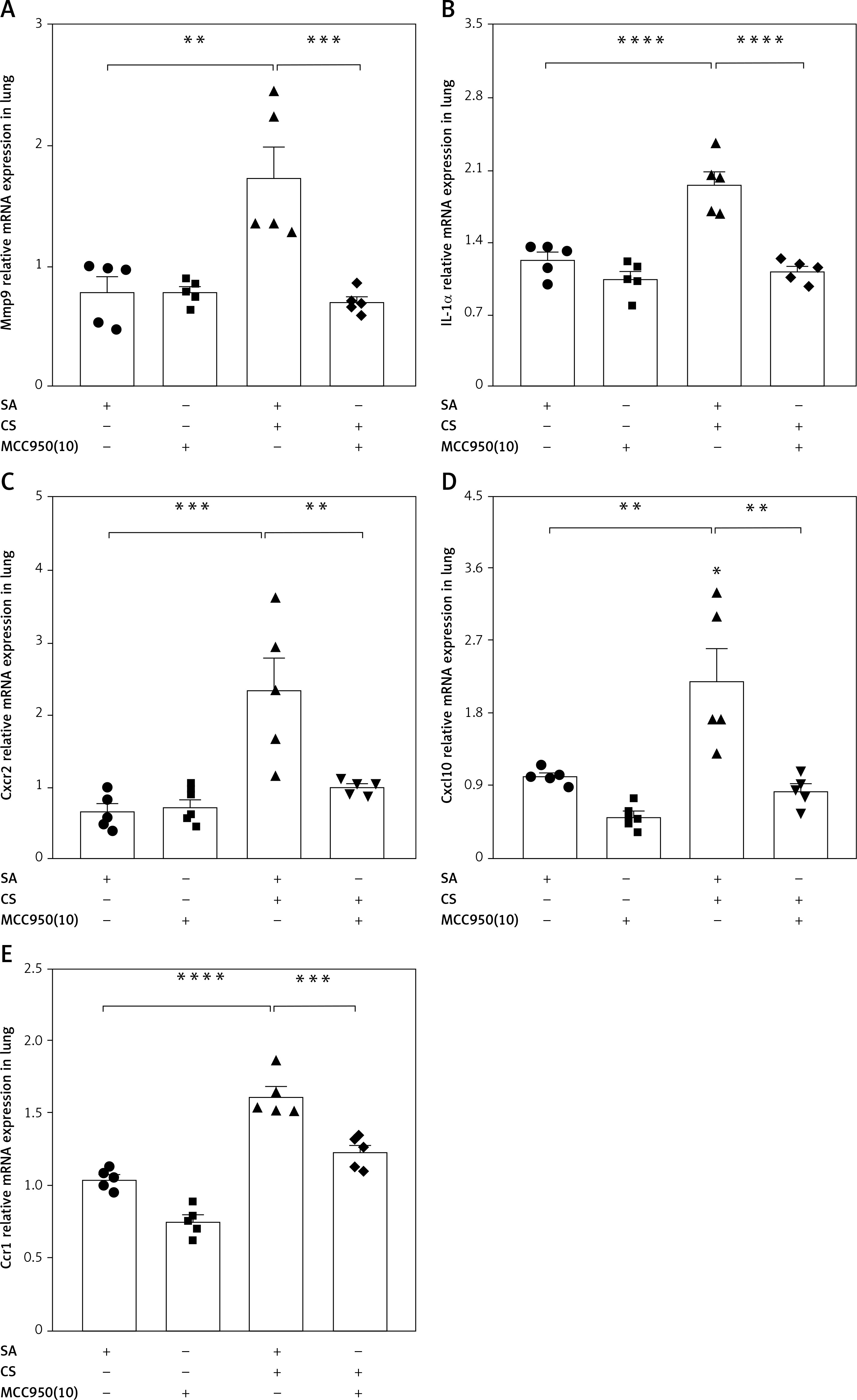
After pharmacological inhibition of NLRP3 by MCC950, 166 genes were down-regulated (CS + MCC950 10 mg/kg vs. CS, |log2FC| > 1, q < 0.05) (Figures 5 B, C and Supplementary Table SII), 84 of which were intersected (Figure 5 C) with the CS-induced upregulated genes (CS vs. SA, |log2FC| > 1, q < 0.05) (Figures 5 A, C and Supplementary Table SII). Then, PPI network analysis was performed with the candidate 84 genes, which indicated that five hub genes, i.e. matrix metalloproteinases 9 (Mmp9), interleukin-1α (IL-1α), CXC motif chemokine receptor 2 (Cxcr2), CXC motif chemokine ligand 10 (Cxcl10), and C-C motif chemokine receptor 1 (Ccr1), located at the center of the PPI network, were most likely to be correlated with NLRP3 (Figure 5 D and Supplementary Table SIII). Moreover, MCODE analysis further implicated that the five hub genes were also assembled in the same cluster with high scores (Figure 5 E and Supplementary Table SIV). Validation with real-time qPCR confirmed that overexpression of all five hub genes induced by CS was significantly alleviated by NLRP3 inhibition (Figure 6 A–E).
Figure 5
The hub genes were identified via bioinformatics analyses. Volcano plots show the log2 fold change (X-axes) plotted against log10-adjusted P values (Y-axes) for (A) CS versus (vs.) SA group and (B) CS + MCC950 (10) vs. CS group, with upand down-regulated DEGs highlighted in blue and green respectively. C – Venn diagram shows an overlap of 84 DEGs (green area) between the two comparisons (red and blue circles). Red circle represents the up-regulated genes of CS vs. SA group, and blue circle represents the down-regulated genes of CS + MCC950 (10) vs. CS group. The Arabic number in brackets represents the dose of MCC950. D – PPI network analyses were performed and visualized using the 84 overlapped DEGs. The bigger circles indicate more related genes and the red circles highlight the hub genes. E – In the MCODE analysis, the top 1 gene clustering module among the 84 overlapped DEGs is presented. The red circles highlight the hub genes
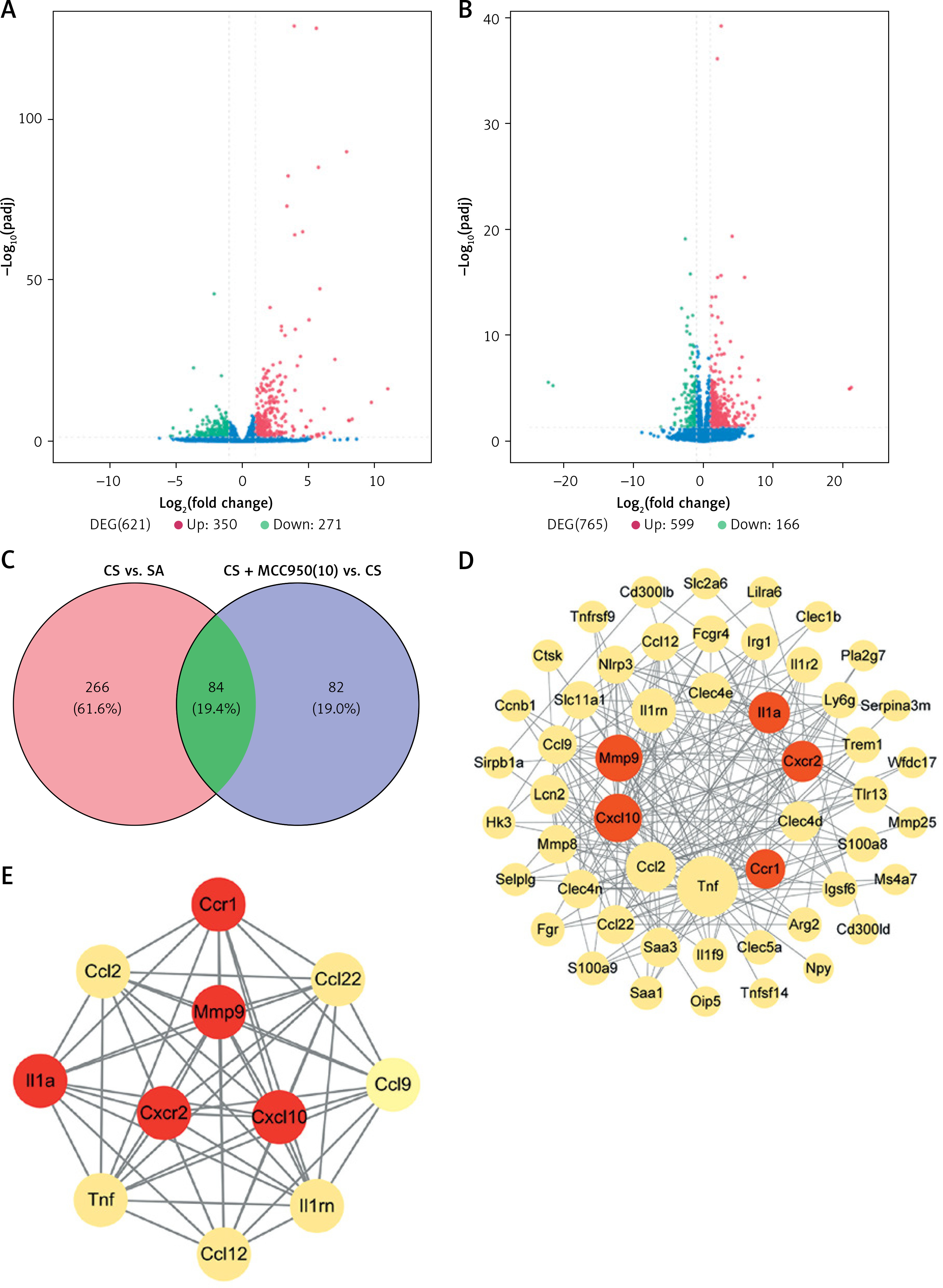
Figure 6
Validation of the hub genes was performed by real-time qPCR. The mRNA expression of the five hub genes Mmp9 (A), IL-1α (B), Cxcr2 (C), Cxcl10 (D), Ccr1 (E), relative to GAPDH, was measured by real-time qPCR. Data are presented as mean ± SEM. The significance of differences was examined by one-way analysis of variance followed by Tukey’s multiple comparisons test. P < 0.05 was considered statistically significant. *P < 0.05, **p < 0.01, ***p < 0.001, ****p ≤ 0.0001
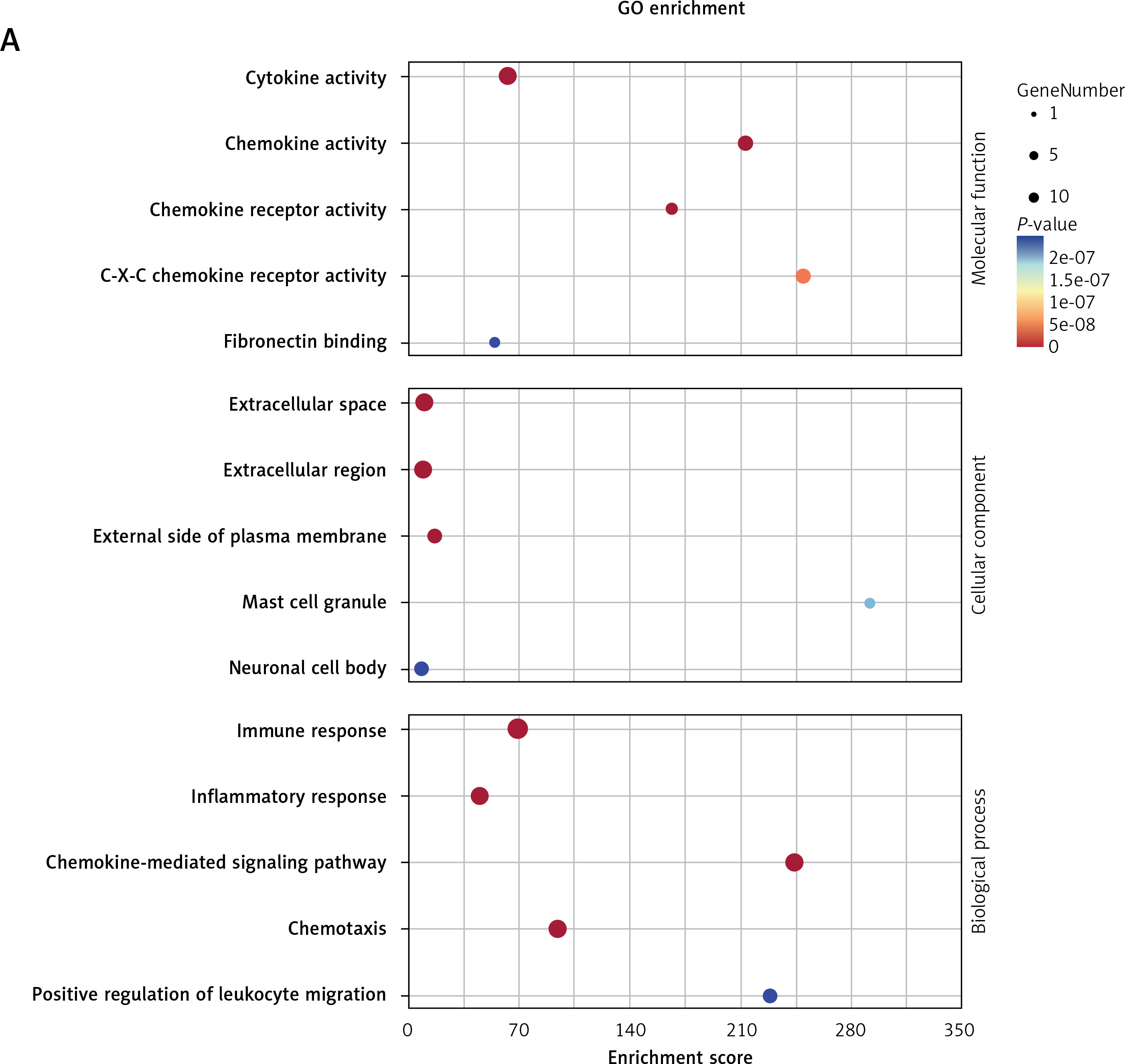
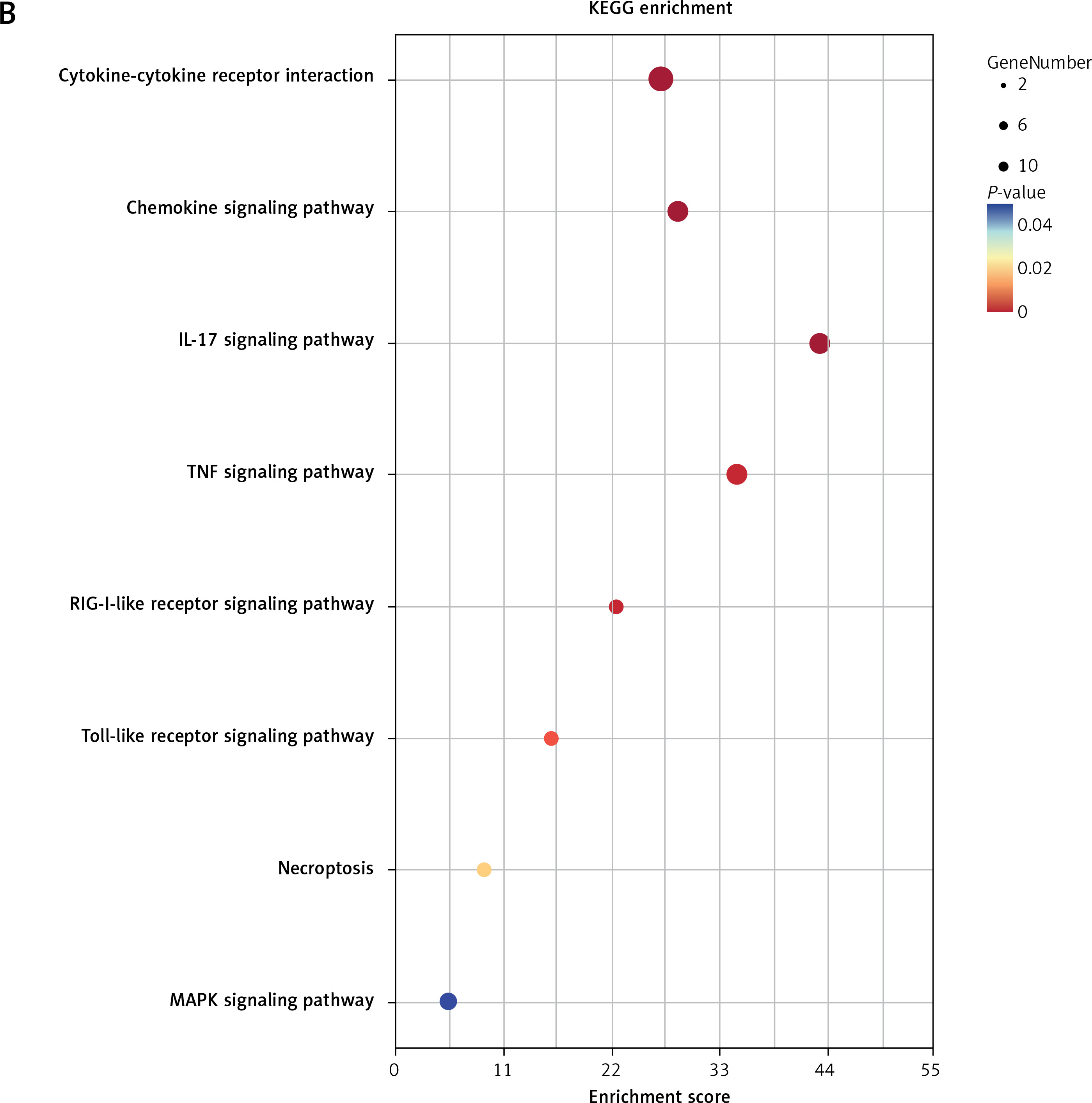
GO and KEGG enrichment analyses further indicated that these five genes were mainly enriched in inflammatory-immune response, cell chemotaxis, and receptor activation in biological processes (Figure 7 A and Supplementary Table SV), as well as toll-like receptor (TLR), RIG-I-like receiver (RLR), interleukins, and tumor necrosis factor (TNF) signaling pathways (Figure 7 B and Supplementary Table SVI).
Figure 7
GO and KEGG enrichment analyses associated with the hub genes. A – Hub gene-related GO terms, including biological processes, molecular function and cell component. A bigger size of the bubble represents more DEGs in the significant GO terms and signaling pathways, and different colors of the bubble represent different p-values. B – hub gene-related signaling pathways in KEGG enrichment analysis are displayed according to the p-values and enrichment scores. A bigger size of the bubble represents more DEGs in the significant GO terms and signaling pathways, and different colors of the bubble represent different p values
Discussion
For end-stage COPD, although lung transplantation is the currently optimal solution [3, 4], stem cell therapy and tissue engineering are promising protocols, due to successful experimental research [24–26]. However, targeted intervention on the critical inflammatory processes in COPD is still the key point, especially in the early stage of the disease. In the present study, we initially reported elevated NLRP3 expression level in the lung tissues and sera of COPD smokers. Moreover, CS-induced airway inflammation in mice was significantly attenuated by pharmacological inhibition of NLRP3 by MCC950 in a dose-dependent manner. Noticeably, five hub genes (Mmp9, IL-1α, Cxcr2, Cxcl10, Ccr1), as well as their related inflammatory processes/signal pathways, which were most likely to be associated with NLRP3, were identified and further validated. These data indicated a positive role of NLRP3 in CS-induced airway inflammation.
An increased level of NLRP3 has been reported in lungs from COPD patients [10]. Our study confirmed that NLRP3 expression was elevated not only in lung tissues but also in sera of COPD smokers, suggesting that activated NLRP3 might contribute to both local (airway) and systemic inflammation induced by CS in COPD. Furthermore, pharmacological inhibition targeting NLRP3 can significantly attenuate LPS or CS or influenza virus induced acute/subacute inflammation [12, 27, 28]. In the present study, 4-week consecutive nose-only CS exposure induced remarkable airway inflammation, which was significantly ameliorated by dose-dependent treatment with MCC950. More importantly, the five hub genes were identified via a series of bioinformatic analyses and subsequently validated by real-time qPCR.
All these five hub genes have been documented to be implicated in inflammatory responses related to NLRP3 and in COPD. Mmp9 expression was reported to be significantly increased in lung tissues of COPD patients [29], which was correlated with NLPR3 activation and emphysema in COPD. However, inhibition of NLPR3 activation could reduce Mmp9 expression in the ischemic retinopathy, ischemic stroke and osteoarthritis rodent models [30–32]. In the present study, the overexpression of Mmp9 induced by CS exposure was downregulated by NLRP3 inhibition, possibly through potential negative effects on regulation of leukocyte migration and signal pathways involved in interleukins [33].
IL-1α, released from damaged epithelial cells, is a trigger for inflammatory responses in lungs [34]. When cultured with CS extract, COPD-derived airway epithelial cells elicited a stronger IL-1α response compared to control-derived airway epithelium [35]. Also, intra-amniotic administration of IL-1α induced activation of the NLRP3 inflammasome in the fetal membranes; however, NLRP3 deficient mice had a reduction in the rates of preterm birth and neonatal mortality caused by the intra-amniotic injection of IL-1α [36]. Similarly, in this study, inhibition of NLRP3 reduced CS-stimulated IL-1α expression associated with airway inflammation, which was implicated in the immune-inflammatory response, such as the mitogen-activated protein kinase (MAPK) pathway [37].
Cxcr2, Cxcl10 and Ccr1 are all chemokine receptor/ligands, deeply participating in immuno-inflammatory responses, especially chemotaxis. Cxcr2 played an important role in neutrophil migration via binding to its specific ligands of Cxcl1 [38], and activated NLRP3 participated in Cxcl1/Cxcr2-induced inflammatory responses [39]. However, inhibition of Cxcr2 could significantly decrease NLRP3 expression and ameliorate neutrophil-dependent airway inflammation [40]. By contrast, our data revealed that inhibition of NLRP3 could attenuate CS-induced overexpression of Cxcr2, suggesting a crosstalk between NLRP3 and Cxcr2 in CS-induced inflammatory responses. Cxcl10 is an essential genetic marker for M1 macrophage activation [41]. It was reported that activation of NLRP3 would promote M1 polarization [42], while blockade of NLRP3 reversed the increase of Cxcl10, inhibiting M1 polarization in TMAO-cultured bone marrow-derived macrophages [43]. In this study, Cxcl10, as well as macrophages in BAL fluid, was upregulated after CS exposure, which was partially reversed with MCC950 treatment, potentially via preventing chemotaxis, cytokine-cytokine receptor interaction, toll-like receptor activation, etc. In addition, Ccr1 was considered to be a diagnostic marker for sputum neutrophil oxidation and airway inflammation in COPD [44]. Our study further indicated that Ccr1 was associated with chemotaxis and cytokine receptor interactions in CS-exposed airway inflammation, and NLRP3 inhibition could obviously downregulate CS-induced Ccr1 expression. Overall, these hub genes played a vital role in NLRP3-mediated airway inflammation induced by CS exposure.
In conclusion, the present study indicated that NLRP3 plays an important role in CS-induced airway inflammation in COPD, which was further elucidated with a group of five hub genes associated with NLRP3 and their inflammatory processes and signaling pathways. These data provide some new experimental evidence for the underlying mechanisms of CS-induced airway inflammation in COPD.