Introduction
Guillain-Barré syndrome (GBS) is a rare autoimmune disorder which attacks the peripheral nervous system, characterized by rapidly progressive symmetrical paralysis in the limbs, sensory dysfunction, and areflexia [1–3]. Experimental autoimmune neuritis (EAN) is a counterpart animal model for studying GBS. To date, the pathogenesis of GBS/EAN has been extensively studied, including cellular and humoral immune responses [4, 5], molecular mimicry, the Th1/Th2/Th17 paradigm [6–9] and Toll-like receptors (TLRs) [10, 11], etc., but is not completely clear.
Toll-like receptors are predominantly expressed on the immune cells, which can induce a wide variety of immune responses to specific pathogens [12, 13]. Increasing evidence suggests that TLRs are involved in the pathogenesis of GBS from different aspects including gene, patients with GBS as well as EAN research. Nyati and Prasad reported that inflammatory genetic polymorphisms may predict risk and susceptibility for development of GBS [14], and they also found that TLR4 (Asp299Gly) polymorphism is related to increased susceptibility to GBS [15]. Du [16] and Wang et al. [10] revealed up-regulated expression of TLR2, 4 and 9 and their relational signaling molecules in patients with GBS. A significant increase of TLR2, TLR6 and TLR11 was observed in the C57Bl/6J EAN mice by Gries [11]. Meanwhile, Deng et al. [17] found TLR4 and TLR9 mRNA expressed highly in Lewis rats with EAN.
Moreover, it is generally accepted that interleukin (IL)-17 plays pivotal roles in autoimmune diseases which facilitates the generation of many chemokines, cytokines, and growth factors. However, whether there is an association of Th17 cells or IL-17 cytokines with TLR4 in EAN remains poorly documented. Wang et al. [18] found that IL-17A participated in development of spontaneous pulmonary emphysema which was caused by Toll-like receptor 4 mutation.
Therefore, to observe the effect of Toll-like receptor 4 (TLR4) deficiency on clinical severity and expression of Th1/Th2/Th17-associated cytokines, we selected C57BL/10 mice and TLR4 knockout (KO) mice with the C57BL/10 background for induction of the EAN model. The results are reported as follows.
Material and methods
Animals
C57BL/10 mice and TLR4 KO mice [19] with the C57BL/10 background were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). Male mice, 4–5 weeks old (18–20 g), were used for the study. All mice were housed in a 12/12 light-dark environment at 25°C in the Animal Care Facility of Jilin University according to institutional guidelines.
Antigen
The P0 peptide 180–199 (SSKRGRQTPVLYAMLD-HSRS) of murine peripheral myelin protein P0 was produced according to the following steps: it was first synthesized by the 9-fluorenylmethoxycarbonyl (Fmoc) solid-phase procedure; then purified by HPLC using a Vydac reverse-phase column (Grace Vydac, Hesperia, CA, USA); and finally analyzed by MALDI-time of flight (TOF) mass spectrometry (Cambridge Research Biochemicals, Billingham, UK).
Induction of experimental autoimmune neuritis and evaluation of clinical signs
We induced EAN by immunizing mice via subcutaneous injection of 180 μg of P0 peptide 180–199 (SSKRGRQTPVLYAMLDHSRS) [20] (GenScript, USA) emulsion in 25 μl of PBS and 0.5 mg of Mycobacterium tuberculosis (Difco, USA) in 25 μl of Freund’s incomplete adjuvant (Sigma Aldrich, USA). On days –1 and +1 after immunization, 400 and 300 ng of pertussis toxin (Merck Millipore, Germany), respectively, was administered intravenously (via tail veins). A blinded protocol was used to assess clinical signs of mice with EAN according to the following criteria: normal (score = 0), reduced tone of the tail (score = 1), flaccid tail (score = 2), abnormal gait (score = 3), ataxia (score = 4), mild paraparesis (score = 5), moderate paraparesis (score = 6), severe paraparesis (score = 7), intermediate clinical signs (score = 0.5). To observe the roles of TLR4 and Th1/Th2/Th17-associated cytokines in the EAN, the animals were grouped as follows: Naive WT group, EAN WT group, EAN TLR4 KO group, which were sacrificed at the peak of clinical EAN, i.e., at day 16 post-immunization. The sera were quickly isolated and stored at –80°C.
Serum cytokine measurement by cytometric bead array
Concentrations of serum cytokines (IL-2, IL-4, IL-6, IL-10, IL-17A, interferon (IFN)-γ and tumor necrosis factor (TNF)) were determined using the Ms Th1/Th2/Th17 CBA kit (BD Biosciences, USA) according to the manufacturer’s instructions. We analyzed beads by flow cytometry using an FACSCalibur flow cytometer and CellQuest software (BD Biosciences, USA). We analyzed data with a spreadsheet developed by BD Biosciences that used a four-parameter logistic curve-fitting model [21].
In our study, all experiments were repeated three times, and the averages were used for statistical analysis. Moreover, our study has been approved by the Ethics Committee of Beijing Tiantan Hospital, Capital Medical University, and the ethical review number is KY2016-052-02.
Statistical analysis
Statistical analysis was performed using SPSS 20.0 for Windows (SPSS Inc., Chicago, IL, USA). All results obtained are presented as mean values ± standard deviation (SD). Differences of the Th1/Th2/Th17 cytokines between the WT mice, the WT mice with EAN and the TLR4 KO mice with EAN were compared using the one-way analysis of variance (ANOVA) or the Mann-Whitney U test. To test for differences of clinical scores between TLR4 KO mice and their WT counterparts, two independent-samples t-test or the Mann-Whitney U test was applied. P < 0.05 was considered statistically significant.
Results
Toll-like receptor 4 deficiency alleviates the clinical severity of experimental autoimmune neuritis
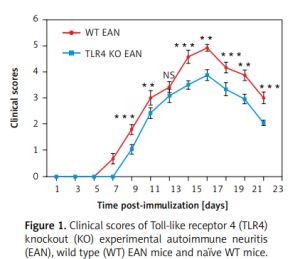
As shown in Figure 1, the onset of EAN in WT mice ranged between days 7 and 8 p.i. In contrast, the TLR4 KO mice experienced a later onset of EAN, i.e., at days 9–10 p.i. From day 9 p.i., the clinical scores of TLR4 KO mice were evidently milder than those of their WT counterparts with the exception of day 13 post-immunization. Differences between them were compared using two independent-samples t-test or the Mann-Whitney U test. **P < 0.01, ***p < 0.001.
Figure 1
Clinical scores of Toll-like receptor 4 (TLR4) knockout (KO) experimental autoimmune neuritis (EAN), wild type (WT) EAN mice and naïve WT mice. Experimental autoimmune neuritis was induced by immunization with P0 peptide 180–199 in combination with pertussis toxin (PTX) and Freund’s complete adjuvant (FCA). All mice received 400 and 300 ng PTX intravenously on days –1 and +1 p.i., respectively. Using a blinded protocol, two different examiners evaluated clinical severity of EAN immediately after immunization (day 0 p.i.) and thereafter every day until day 24. After 24 days p.i., the clinical course began to recover. Results from the three groups are presented as mean values ± SD (n = 6 in every group). From day 9 p.i., the clinical signs of TLR4 KO mice were significantly milder than those of their WT counterparts with the exception of day 13 post-immunization. Differences between them were compared using two independent-samples t-test or the Mann-Whitney U test. **P < 0.01, ***p < 0.001, NS – no significance
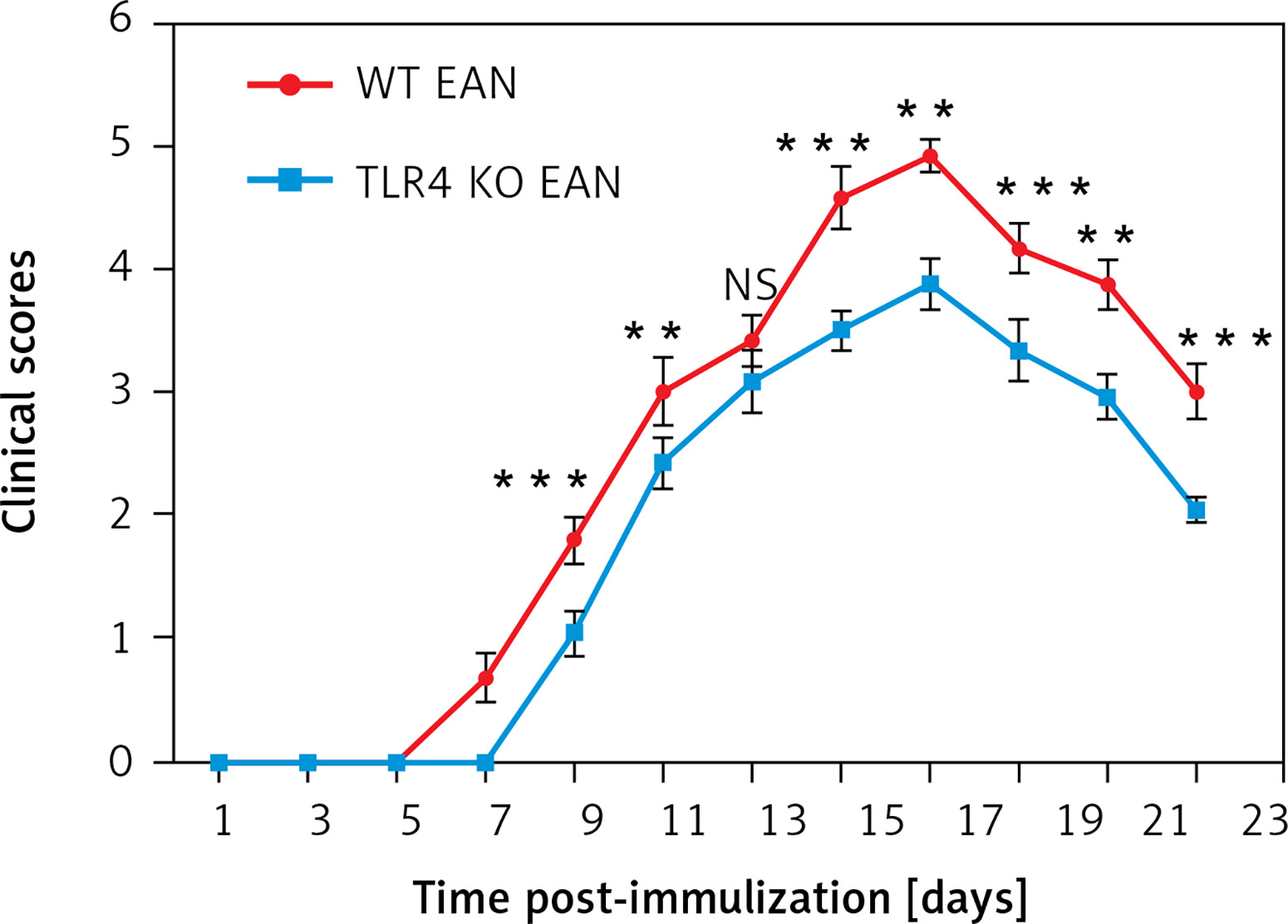
Toll-like receptor 4 deficiency decreases the production of IL-17A
To prove the involvement of serum cytokines in the pathogenesis of EAN, mice were sacrificed at the peak period of disease (day 16 p.i.) and the sera were collected rapidly for Th1/Th2/Th17 cytokine (including IL-4, IL-6, IL-10, IFN-γ, TNF, IL-17A) detection by cytometric bead array (CBA). Compared to the WT mice, the WT mice with EAN showed significant elevation in IL-6, IL-17A, IFN-γ and TNF. However, there were no significant differences in IL-4 and IL-10 concentrations between them. Furthermore, the results suggest that only sera IL-17A was statistically significantly higher in WT mice with EAN than that of their TLR4 KO counterparts (p < 0.05), but there was no difference in IFN-γ, TNF, IL-4, IL-6, IL-10 concentrations between them (Figure 2). Differences between them were compared using the one-way analysis of variance (ANOVA) or the Mann-Whitney U test. *P < 0.05, **p < 0.01, NS – no significance.
Figure 2
Expression of interleukin (IL)-4 (A), IL-10 (B), IL-6 (C), IL-17A (D), interferon (IFN)-γ (E) and tumor necrosis factor (TNF) (F) of sera from naïve wild type (WT), WT experimental autoimmune neuritis (EAN) and Toll-like receptor 4 (TLR4) knockout (KO) EAN mice. Mice were sacrificed at the peak period of disease (day 16 p.i.) and the sera were collected rapidly for Th1/Th2/Th17 cytokine detection by cytometric bead array (CBA). Data are represented as means. Except for IL-4 and IL-10, the expression of IL-6, IL-17A, IFN-γ and TNF was significantly higher in WT EAN mice compared with naïve WT mice. However, only the expression of IL-17A was significantly higher in the WT EAN mice compared with the TLR4 KO EAN mice. Differences between them were compared using two independent-samples t-test, the Mann-Whitney U test or one-way analysis of variance (ANOVA). **P < 0.05, **p < 0.01, NS – no significance
Discussion
At present, the GBS animal model of EAN, which can mimic the human GBS, is used widely. In our study, we induced EAN in C57BL/10 and TLR4 KO mice. We found that the onset of EAN in TLR4 KO mice was significantly delayed. Meanwhile, we noted a trend of milder clinical signs of EAN in TLR4 KO mice as well. Further, we found that TLR4 deficiency only reduced the production of Th17 cytokine IL-17A, but did not downregulate the expression of other Th1/Th2/ Th17-associated cytokines, such as IL-4, IL-6, IL-10, IFN-γ, and TNF. Consistently with our study, Zhang et al. [22] found that IL-17(+) cells might contribute to the pathogenesis of EAN by examining the spatiotemporal expression of IL-17 using immunohistochemistry and RT-PCR, and analyzed the IL-17(+) cell proportion in blood and lymph nodes using flow cytometry. Combined with our data, we suppose that TLR4 deficiency may mitigate experimental autoimmune neuritis via modification of the Th17/IL17 responses. The exact conclusion needs to be explored further, such as detecting the spatiotemporal expression of IL-17 using immunohistochemistry or RT-PCR in TLR-4 mice.
Additionally, the study revealed that reduced expression of IL-6, IL-12p40, and IL-17A in nuclear translocation of RelA EAN knockout mice and obviously elevated Tregs were found in TLR4 KO mice as compared to wild-type mice in the recovery phase. A study found that cytokines such as IL-6, IL-17A, Th/Tregs and TLRs may be involved in the disease progression [23]. However, our research indicated that there may be a link between TLR4 and IL17/Th17 in the pathogenesis of GBS.
Generally, researchers suggest that a disturbance of the cytokines Th1/Th2/Th17/Treg balance is one of the main pathological mechanisms in GBS or EAN [9, 24]. Li et al. proved that the amount of Th1/Th2/Th17 cells was obviously increased in GBS [24]. Given the important roles of cytokines in the immunopathogenesis in both GBS and EAN, we sought to examine a broad spectrum of serum cytokines in WT and TLR4 KO mice. In line with investigations by Matsui et al. [25], we observed that the serum expression of Th1 cytokines IFN-γ, TNF, IL-6 and Th17 cytokine IL-17A were increased in WT EAN mice while expression of Th2 cytokines IL-4 and IL-10 was not significantly different when compared with WT mice. Similar results were also obtained in other studies [26, 27]. However, Zhang et al. observed aggravated clinical scores despite an alleviated systemic Th1 immune response in IFN-γ deficient mice with EAN [28]. Interestingly, our current data showed that serum IL-17A and clinical score were lower in TLR4 KO mice with EAN than in their WT counterparts, which indicated a certain link between the TLR4 signaling pathway and Th1/Th2/Th17 responses in C57BL/10 mice with EAN. Some studies demonstrated that the TLR4 mRNA is highly expressed on Th17 cells and the TLR4 signaling pathway increases Th17 activation and persistence but weakens Th1 responses [29, 30]. Moreover, Xu et al. [31] reported that TLR4 signaling inhibited Th1 cell differentiation by suppressing the STAT1 pathway and facilitated Th17 cell differentiation by activating the STAT3 pathway in the mouse model of MPE. Increasing evidence shows that TLR4 activation plays a critical role in the development of Th17cells and the expression of Th17-associated cytokines under normal or infectious situations [29, 32, 33]. Bhan and his team showed that Gam-negative bacterium induced the expression of IL-17 by activating TLR4 [34]. The attenuation of IL-1β, TNF-α, IFN-γ, IL-17, and IL-23 was found in the lungs of TLR4-mutant mice after vaccination with pertussis vaccines [35].
In conclusion, TLR4 deficiency could attenuate the clinical severity and delay the onset of EAN. Meanwhile, it also reduced the production of the inflammatory cytokine IL-17A. Based on these findings, TLR4 may contribute to the pathogenesis of EAN by regulating Th17 cells and the level of Th17-associated factors. However, the exact mechanism remains unclear and more evidence is needed to elucidate its role in EAN.