Introduction
Alzheimer’s disease (AD) is one of the most widespread and the most comprehensively studied neurodegenerative disorders. AD pathogenesis has been mainly attributed to extracellular aggregates of amyloid β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) made of hyperphosphorylated τ-protein in cortical and limbic parts of the human brain [1].
Mutations in the genes encoding amyloid precursor protein (APP) and presenilin 1 and 2 (presenilin 1 gene, PSEN1; presenilin 2 gene, PSEN2) are associated with Alzheimer’s disease and lead to an increase in the production of toxic Aβ [2, 3]. Additionally, the mutations in the PSEN1 and PSEN2 genes increase the production of an amyloidogenic form of Aβ such as Aβ42 [2]. It was also shown that the absence of presenilin expression in the neurons resulted in decreased NMDA receptor levels in the synaptic membrane and altered CREB/CBP gene regulation in the neuron, resulting in impaired BDNF expression [4].
Neurofibrillary tangles are another major pathological feature of AD. Under pathological conditions, abnormal phosphorylation of intracellular tau protein leads to the loss of its biological activity and causes microtubule depolymerization, which leads to degeneration of neurons [1]. Other proteins, such as apolipoprotein E (APOE), also play an important role. It was shown that the C-terminal fragments of protein APOE also can bind to Aβ, causing the growth of amyloid deposits. The ε2 (APOEε2) and ε3 (APOEε3) isoforms are mainly involved in the repair of neurons in the peripheral and central nervous system, and, in addition, play a major role in regulating the metabolism and distribution of cholesterol in neuronal membranes. However, the ε4 isoform (APOEε4) is associated with the intensification of amyloidogenic processes. The occurrence of 2 copies of the APOEε4 allele increases the risk of developing AD 16-fold [5].
The development of neurodegenerative disorders such as AD may also be influenced by age-related changes in innate immunity, which provides the first line of defence by recognizing pathogen-associated microbial patterns, inducing key costimulatory molecules and cytokines that activate the adaptive immune response [6].
Current pharmacotherapy used during Alzheimer’s disease is symptomatic and is administered to slow the disease progression and reduce the severity of behavioural disorders. The treatment strategy involves the use of cholinesterase inhibitors (donepezil, rivastigmine) and an N-methyl-D-aspartate receptor (NMDA receptor) antagonist (memantine) [7, 8]. The disease is primarily characterized by memory loss and decline of other cognitive functions, involving those associated with abstract reasoning, language difficulties, agnosia, and disorientation, but also behavioural or personality disorders, which affect the quality of life of both the patient and their loved ones. The illness significantly impacts the patient’s daily professional activities and social functioning. Hence, it is a serious health problem for patients and their families as well as the healthcare system [7, 9]. Projections assume that in the face of a rapidly aging population, there will be 80 million people worldwide suffering from dementia by 2040 [10]. Thus, research on preventive and therapeutic measures is warranted. Previous research on therapeutic strategies has focused on the amyloid cascade hypothesis. However, there is new research into the therapeutic effect of neuroprotective, anti-inflammatory, antioxidant, immunotherapeutic, chelating, and amyloid binding agents as well as secretase modulators [7]. This study presents the relationship between cystatin C (acquired from egg white) and neurodegenerative diseases, indicating its potential use in supporting cognitive functions.
Classification of cystatins
In 1981, Alan J. Barrett first used the term “cystatin” to describe a protein isolated from egg white that could inhibit the activity of lysosomal cysteine proteases [11]. The cystatins are a group of proteins typically composed of 100–120 amino acids [12]. They are papain-like cysteine protease inhibitors (C1) and some of them, like ovocystatin, also inhibit the Leguminosae family (C13) [11, 13].
Cystatin superfamily
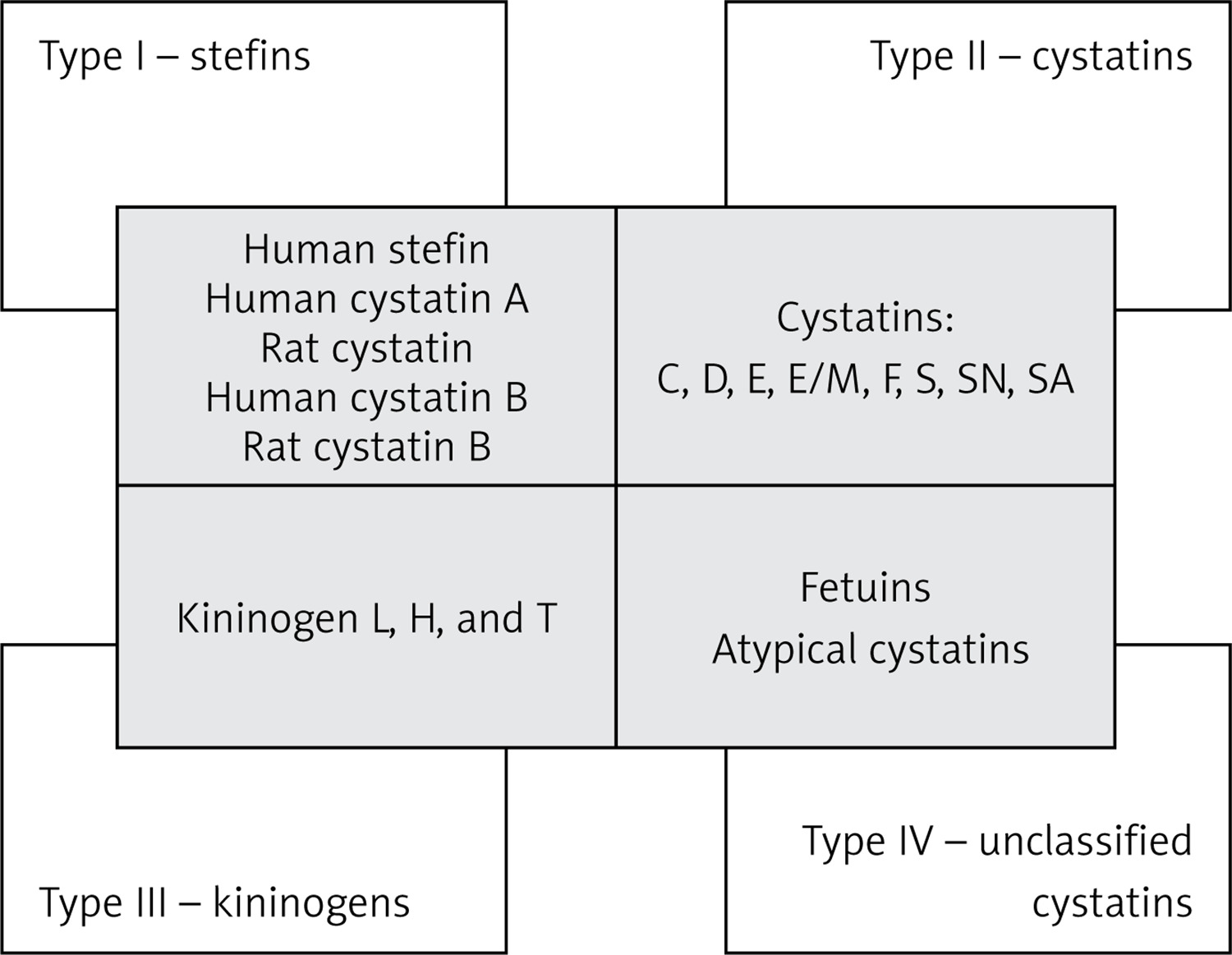
The cystatin superfamily is divided into 4 types/families based on their characteristics as well as their presence in the intracellular, extracellular, and intravascular compartments (Figure 1). Type 1 cystatins, also named stefins, are mainly intracellular, unsynthesized cystatins that are pre-proteins containing signal peptides. Proteins representing this family are named “human stefin”, “human cystatin A”, “rat cystatin”, “human cystatin B”, and “rat cystatin B”. They contain about 100 amino acid residues (~11 KDa) and lack disulphide bonds and carbohydrate side chains [14]. Stefin B has been shown to have an anti-inflammatory character via regulation of caspases and NF-κB activation, and also to regulate lysosomal proteolytic activity and therefore intracellular clearance [15, 16]. Type 2 mainly contains cystatins secreted extracellularly, such as cystatins C, D, E, E/M, F, S, SN, and SA. They possess about 120 amino acid residues (~14 KDa) and 2 conserved disulphide bonds at the carboxyl terminus. Type 3 cystatins, also known as kininogens, include low- (50–68 kDa) and high- (88–114 kDa) molecular-weight intravascular cystatins, which are mainly plasma proteins and contain 3 cystatin domains [11, 13, 17]. They are synthesized in the liver in the form of multidomain glycoproteins [18]. It was also shown that kininogens are involved in both innate and adaptive immune responses [19]. The type 4 group, containing fetuins and unclassified “atypical” cystatins, is distinguished in some classifications [11, 20, 21]. Another classification system is based on the sequence similarity and 3D spatial structure of the proteins. In this classification, the cystatins belong to the I25 family, with a further division into 3 subfamilies: I25A, I25B (where ovocystatin was classified), and I25C [22] (Figure 1).
Cystatin C
The first studies on cystatin C date back to 1961, when Jorgen Clausen first described the presence of a protein in the cerebrospinal fluid, which he called γ-CSF (γ-cerebrospinal fluid) [11]. In the following years, its presence was reported in cerebrospinal fluid, urine, and abdominal and pleural effusions. Due to the γ-electrophoretic mobility, it was called the “γ-trace” or “post-γ protein”. The final name “cystatin C” was established in 1984 by Alan J. Barret et al. [11, 20].
Structure and function of cystatin C
Cystatin C is an endogenous inhibitor of cysteine proteases and is classified as a type 2 cystatin. The active and mature form of human cystatin C is a non-glycosylated polypeptide chain with a molecular weight between 13.343 and 13.359 kDa. It is composed of 120 amino acids with 2 disulphide bonds: Cys 73-83 and Cys 97-117 [11, 20, 23–25]. It is monomeric both in vivo and in vitro. However, it may be converted into a dimer in elevated temperatures, at a low pH, and by using chemical denaturation [12]. Cystatin C also undergoes 3D domain swapping, which can lead to the formation of amyloid plaques [11, 12, 17]. Human cystatin C is encoded by the CST3 gene, located on chromosome 20 (20.p11.2), consisting of 3 exons and 2 introns [23, 26]. Its presence has been confirmed in all mammalian tissues and fluids, including the cerebrospinal fluid, blood plasma, semen, urine, and saliva [24, 25, 27]. It is also expressed in neurons, astrocytes, microglial cells, and the choroid plexus. Its presence in brain tissue is eminent [28, 29], and its concentration in the cerebrospinal fluid (CSF) is 5 times higher than in the blood [30]. Cystatin C has primarily been studied in the context of renal dysfunction and as a biomarker of the glomerular filtration rate (GFR). However, it has a much wider biological role involving its antibacterial and antiviral properties, a role in bone resorption, tumour metastasis, modulation of the immune system, and proliferation and cell growth [25, 28]. Brain cystatin C levels increase following injury, such as ischaemia, axotomy, or surgery [29]. The blood levels of cystatin C may increase in the course of aging, chronic nephritis, neoplasia, urinary tract infections, hypertension, cardiovascular diseases, rheumatoid arthritis, and glucocorticoid pharmacotherapy [27, 28, 31]. As an endogenous inhibitor of cysteine proteases, cystatin C inhibits cathepsins B, H, L, and S [29, 32]. An imbalance between the proteases and inhibitors may result in the development of pathological conditions favouring excessive proteolysis and leading to abnormal tissue morphology or neuronal cell death [23, 33, 34].
The cystatin C gene and neurodegenerative diseases
Since human cystatin C was found to coexist with β-amyloid depositions, multiple studies have focused on the relationship between the CST3 gene and neurodegenerative diseases. Cerebral amyloid angiopathy (CAA) is characterized by the deposition of amyloid plaques in the vascular wall of arteries, arterioles, and, less commonly, capillaries and veins. The Icelandic type of CAA, known as hereditary cystatin C amyloid angiopathy (HCCAA) or hereditary cerebral haemorrhage with amyloidosis of Icelandic type (HCHWA-I), leads to a haemorrhagic stroke and to the development of dementia. HCCAA is caused by a point mutation in the CST3 gene (Leu to Gln mutation at position 68, Leu68Gln, L68Q) [28, 35]. The cystatin C gene has 3 polymorphic sites in the 3’ region. The G/A polymorphism (G73A) of the CST3 gene in the +73 position leads to an alanine to threonine substitution in the region encoding the signal peptide. It should be mentioned that the polymorphism responsible for encoding alanine is known as allele A, while the one responsible for the expression of threonine in the same position is known as allele B [36–38]. The study by Finckh et al. [29], carried out on AD patients and controls ±75 years old showed that haplotype B of the CST3 gene is associated with late-onset Alzheimer’s disease, regardless of APOEε4. Moreover, the study by Crawford et al. [39] suggests that the CST3-A gene may be a risk factor for late-onset Alzheimer’s disease, also regardless of the APOEε4. However, Cathcart et al. [40] showed that the CST3-A and APOEε4A gene interaction led to a 14-fold increased risk of AD in men and a 16-fold increased risk in women. Additionally, Beyer et al. [41] suggested that the CST3-A gene may be a risk factor in early-onset Alzheimer’s disease. Furthermore, a synergistic association between the CST3-A and APOEε4 gene, and AD was found in patients with Alzheimer’s disease between 60 and 74 years old. Another cystatin C polymorphism (Ala25Thr) is associated with both late-onset Alzheimer’s disease and macular degeneration and leads to alternative signal sequence cleavage. In consequence, the altered cystatin C is prone to aggregate formation and loses the inhibitory function of the protein in relation to both β-amyloid and cysteine proteinases [42, 43].
However, some studies provide mixed findings about the association between these genes and AD. Namely, Maruyama et al. [44] did not find any association between the CST3 gene polymorphism and Alzheimer’s disease. A similar finding was reported in the Italian population, where there was no relationship between AD and CST3 either alone or in conjunction with APOEε4 [45]. In addition, a clinical control study also showed no association between the CST3 gene, age, APOEε4, and Alzheimer’s disease [37]. Nevertheless, a study conducted on the Chinese population showed that, although the CST3A allele frequencies were comparable in AD and the control group, the CST3A/A homozygote was significantly associated with late-onset AD [46]. Other studies on the Chinese population revealed the associations between CST3B/B genotype and AD patients older than 75 years, or VD patients younger than 75 years. Moreover, both CST3 and APOEε4 genes were shown to be risk factors for vascular dementia [47]. Research into the Finnish population, carried out by Helisalmi et al., did not find a link between the CST3 gene and the risk of Alzheimer’s disease [48]. Considering the inconsistent results of studies in this field, including those mentioned above, Hua et al. carried out a meta-analysis of the relationship between the cystatin C G73A polymorphism and Alzheimer’s disease. The analyses indicate that this association is present in the Caucasian population and is absent in the Asian population. However, it has to be mentioned that the overall studied Asian population was smaller than the Caucasian one [49]. Babiloni et al. revealed the relationship between AD genetic risk factor and electroencephalography results in which the α 1 and α 2 amplitudes were statistically larger in patients with the B haplotype of the CST3 gene than controls [36]. Maetzler et al. showed that the BB genotype of the CST3 gene is associated with low cystatin C levels in the cerebrospinal fluid in patients suffering from Lewy body dementia [50]. Furthermore, the relationship between cystatin C and presenilin 2 was also found. Namely, 2 mutations of the presenilin 2 gene (PS2 M239I and T122R), linked to familial Alzheimer’s disease, result in a reduction of extracellular secretion of cystatin C [51].
Cystatin C as a biomarker
Previous studies have attempted to determine the relationship between cystatin C levels in cerebrospinal fluid and blood and cognitive impairment or Alzheimer’s disease. The level of cystatin C in the cerebrospinal fluid was found to decrease in patients with AD [52]. On the other hand, higher CSF cystatin C concentrations correlated with more aggravated hippocampal and whole-brain atrophy [53]. In addition, amyloid β1-42 was found to colocalize with cystatin C in patients with AD. This suggests that a decreased level of cystatin C in the CSF may be associated with increased deposition of amyloid peptides [30, 52]. Moreover, cystatin C CSF levels positively correlate with the levels of tau protein and its phosphorylated form, regardless of the APOE genotype, gender, or age of the patient [54]. However, little is currently known about the possible biological interactions between the levels of tau protein and cystatin C. Probably, the expression and secretion of cystatin C increases in response to increased neurodegenerative process [24]. Considering the data on cystatin C binding with β-amyloid, it may also be considered as an attempt to limit amyloid aggregation [55]. CSF cystatin C levels were also compared in patients with AD and vascular dementia, and they were higher in the latter group [56, 57].
So far, studies assessing blood cystatin C levels provide conflicting results. Research by Kálmán et al. [56] showed that, despite a higher blood cystatin C level in patients with vascular dementia, it was not significantly different from that in the group of AD patients and the control group. Yaffe et al. also analysed the connection between cystatin C and cognitive impairment, indicating that high serum cystatin C levels are associated with cognitive impairment in the elderly [58]. Recently, the study of Cui et al. also demonstrated that higher blood levels of cystatin C were positively associated with the cognitive impairment in middle-aged and older Chinese people. Additionally, respondents with higher levels of cystatin C had lower scores on cognitive tests [59]. Consistent with these results, Higgins et al. observed that elevated serum cystatin C was associated with 1.2-fold higher prevalence of dementia in a large, diverse sample of middle-aged and older United States adults [60]. Similarly, obese patients undergoing bariatric surgery with elevated cystatin C serum levels obtained lower scores in a neurocognitive assessment prior to the surgery. However, the follow-up improvements in cognitive performance were independent of the observed normalization of cystatin C concentrations [61]. There has also been a report that found plasma cystatin C levels to be lower in AD patients than in controls [46]. However, long-term studies carried out on a large group of patients led to the conclusion that low levels of serum cystatin C precede clinically manifest Alzheimer’s disease [62]. A longitudinal analysis by Slinin et al. indicates that the relationship between cystatin C and cognitive decline may be U-shaped, with both very low and very high serum cystatin C concentrations predictive of impaired future cognitive performance [63].
At present, there are no means of identifying the development of the disease from mild cognitive impairment (MCI) to Alzheimer’s disease. Cystatin C may prove to be a prognostic factor. Ghidoni et al. suggest that a low plasma cystatin C level may be an important indicator of the conversion of mild cognitive impairment to Alzheimer’s disease [64]. Therefore, cystatin C was the subject of multiple studies as a potential biomarker or diagnostic tool to identify cognitive impairment and Alzheimer’s disease [58, 65, 66]. Reports by Mares et al. [67, 68] imply that cystatin C will not be used in differential diagnostics, although it was initially thought that it could play a major clinical role [69].
The relationship of cystatin C and β-amyloid
Cystatin C co-deposits with amyloid-β in AD patients’ interstitial, neuropil, and brain blood vessels, as well as in pyramidal neurons located in the III and IV layer of the cerebral cortex [70]. Cystatin C could also be localized in the entorhinal cortex, hippocampus (particularly the CA1 area), the temporal cortex, and, to a lesser extent, in the pyramidal neurons of the frontal, parietal, and occipital lobes, including those in the II layers, which was indicated in immunostaining studies. Moreover, together with cathepsin B, cystatin C was reported to be present in cortical glial cells, endosomes, and lysosomes in the brains of patients with Alzheimer’s disease [71]. Cystatin C and amyloid-β co-deposition was also confirmed in the brains of the Rhesus monkey (Macaca mulatta) and Saimiri (Saimiri sciureus) [72]. It has been determined that an overexpression of human cystatin C in the brains of APP transgenic mice reduces β-amyloid deposition and inhibits plaque formation [73]. Both in vivo and in vitro studies confirmed the binding of cystatin C with soluble amyloid-β. In addition, the inhibition of amyloid-β deposition in transgenic mice with an amyloid precursor protein (APP) mutation were shown [31]. It should be mentioned that the overexpression of human cystatin C does not influence the level of the endogenous murine amyloid-β in the brain [74], or the APP expression. Nevertheless, it might reduce the accumulation of β-amyloid in APP/CysC mice compared to APP mice [31]. Steinhoff et al. showed that APP expression in transgenic mice was also associated with an increase in the accumulation of cystatin C in astrocytes, throughout the brain, independently, and before the onset, of amyloid plaque formation [75]. Some findings showed the association of cystatin C and Aβ demonstrated a specific, saturable, and high-affinity binding between cystatin C and both Aβ1-40 and Aβ1-42. Notably, cystatin C association with Aβ results in a concentration-dependent inhibition of Aβ fibril formation [76, 77]. It seems that especially the oligomeric form of cystatin C has a potent role in reducing β-amyloid aggregation, possibly due to some similar structural features [55]. Post-mortem studies conducted on patients with Alzheimer’s disease and controls revealed that human cystatin C binds to soluble β-amyloid. [78]. Additionally, in vitro studies indicate a protective effect of cystatin C on rat hippocampal neurons against the toxic effect of oligomeric or fibrillar forms of β-amyloid [79].
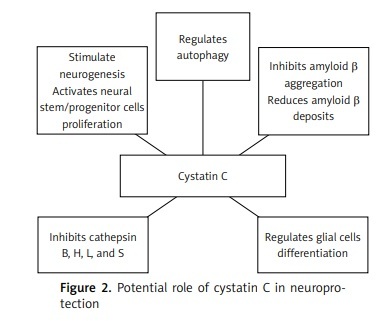
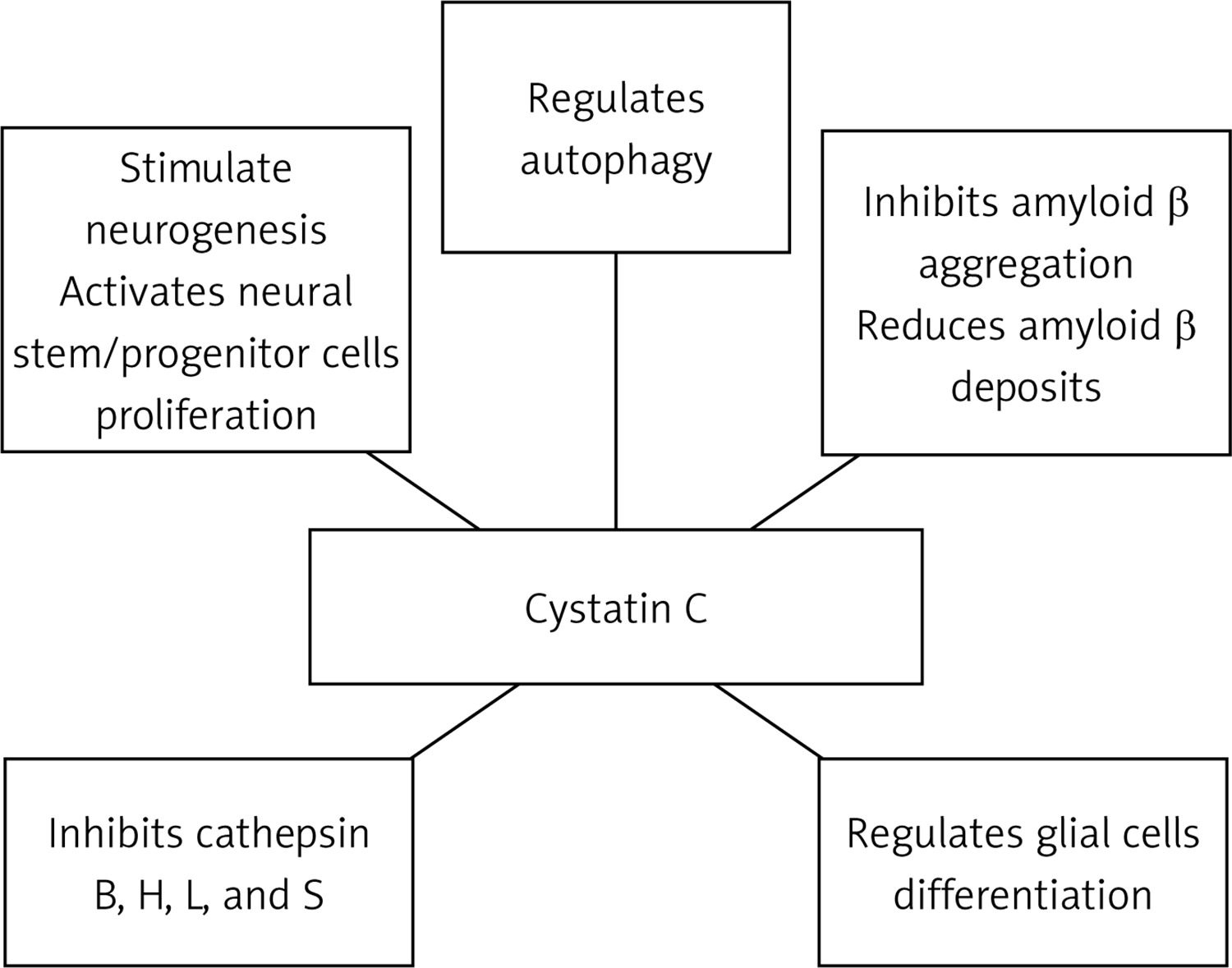
Some evidence demonstrated that cystatin C might participate in the protection processes against neurodegeneration, not only by inhibition of the oligomerization and formation of amyloid-β fibrils, but also by inhibition of cysteine proteases, and induction of autophagy and neurogenesis [25, 28]. In vitro studies have shown that cystatin C inhibits B, H, K, L, and S cathepsins [25, 28]. Cathepsins B and D were found in senile plaques of patients with Alzheimer’s disease [80]. Cathepsin B has also been found to coexist with cathepsin C in these patients [71]. It has been shown that cathepsin B affects the formation of amyloid-β, possibly by enhancing the activity of β-secretase [81]. Similarly, Hook et al. indicated that cathepsin B may act similarly to β-secretase. Therefore, inhibiting its activity reduces the formation of amyloid-β in the course of Alzheimer’s disease [82]. The use of cathepsin B inhibitors in an in vivo study on transgenic mice with the London APP mutation showed a reduction of memory deficits, amyloid-β in the brain, and a decreased activity of β-secretase. These changes were not observed in the Swedish/London APP mice [83]. Interestingly, increased activity of cathepsin B and D was observed in CysBKO mice. However, decreased cathepsin B activity compared to CysBKO mice occurs in CysBKO/CysC mice (a cross of homozygous CysBKO mice and mice with CysC-overexpression). Thus, the overexpression of cystatin C in CysBKO mice may act as a neuroprotective agent by inhibiting cathepsin activity [84]. Nevertheless, some reports of the anti-amyloid and neuroprotective effect of cathepsin B are in contrast to the above-described reports of its role as a cystatin C inhibitor [85, 86].
The neuroprotective functions of cystatin C
Autophagy is one of the main processes of degradation of long-lived proteins and organelles, and it is essential for the survival of neurons. Autophagic malfunction has been suggested to be an element of the pathogenesis of neurodegenerative disorders, including Alzheimer’s disease [87]. Tizon et al. showed that cystatin C induces autophagy and could be considered as a neuroprotective agent [88]. Autophagy activated by cystatin C is manifested as an increase in the degradation of long-lived proteins, which might prove its direct neuroprotection. Moreover, cystatin C showed neuroprotective properties in cell cultures in conditions of cytotoxic changes, where there is nutritional deprivation, colchicine or staurosporine deficiency, or oxidative stress [88]. Under conditions of stress, elevated cystatin C levels induced autophagy by inhibiting the activity of mTOR, which is a negative regulator of autophagy. When autophagy was impeded by 3-methyladenine, an inhibitor, cystatin C no longer had protective functions against the toxicity of nerve cells [88]. It has been demonstrated that the silencing of the gene expression of the Beclin-1 protein, which is an autophagy activator, by siRNA (small interfering RNA) also eliminated the protective effect of cystatin C on serum-deprived cells undergoing apoptosis [88].
Reports from the 1980s and 1990s describe the involvement of cystatin C in the regulation of cell proliferation [89, 90]. Hu et al. demonstrated that the secretion of cystatin C mediates APP-induced neural stem/progenitor cell (NSPC) proliferation [91]. Together with fibroblast growth factor 2 (FGF-2), glycosylated cystatin C also affected the stimulation of neurogenesis in the dentate gyrus of the hippocampus of adult rats [92]. The role of cystatin C in neurogenesis has also been confirmed in cystatin C knockout mice, where its basic levels were decreased in the subgranular zone of the dentate gyrus of the hippocampus [93]. In addition, cystatin C can be involved in the process of astrocytic differentiation during brain development in mice [94], and it can affect the differentiation of embryonic stem cells into neural stem cells [95]. The effect of cystatin C on the development of glial cells, including astrocytes, has been confirmed by Hasegawa et al. [96].
Interestingly, research shows that various psychiatric drugs may interact with cystatins, leading to their decreased availability and anti-proteolytic function [34]. While the significance of this phenomenon remains unknown, it could also contribute to the cooccurrence of mental disorders such as schizophrenia, bipolar disorder, or depression with Alzheimer’s disease [97].
Taken together, the numerous functions of cystatin C make it a candidate for the development of pharmacological therapeutic interventions for neurodegenerative diseases, such as Alzheimer’s disease [98] (Figure 1). However, due to the high costs of the production, the current focus has turned to natural cystatins, such as ovocystatin (Figure 2).
Egg white cystatin – ovocystatin
Egg white cystatin, or ovocystatin, is a type 2 cystatin. It is the most well-known and first isolated protein of the cystatin superfamily [11, 99, 100]. It serves as an inhibitor of the C1 group of cysteine peptidases, including the B, H, K, L, and S cathepsins [33, 99]. Chicken cystatin has very similar biological functions to human cystatin C, which stems from its structure similarities. The amino acid sequence of Ovocystatin is 44% homologous to human cystatin C, whereas there is 62–63% structural homology between the proteins. There is also a strong similarity between the secondary structures of the proteins in solution and in crystalline form [11, 23, 101]. The protein build of ovocystatin comprises a 5-stranded anti-parallel β-sheet wrapping around a central-α-helix, and the potential site of β-amyloid binding is the C-terminal hydrophobic sequence formed by amino acids 99 to 115. For cystatin C, the site with the highest β-amyloid binding affinity resides at the analogous structure within the L2 loop and β5 strand of the protein formed by amino acids 101–117 [102, 103].
The molecular weight of the chicken cystatin is 13.147 kDa, and its single polypeptide chain is comprised of 116 amino acids. There are 2 forms of ovocystatin: a phosphorylated (pI = 5.6) and an unphosphorylated form (pI = 6.5) [104, 105]. Ovocystatin has a high pH and is thermostable. It loses its inhibitory activity in the process of freezing or lyophilization. This can be deterred in formulations with a concentration of less than 10 µM by adding 20% of glycerol [33, 99, 100]. Chicken egg white cystatin undergoes the 3D swapping phenomenon, which is a mechanism for forming dimers or higher oligomers by exchanging protein domains that remain covalently connected to the core domain through the polypeptide chain [106] and can form aggregates. In this process, it undergoes spontaneous dimerization, followed by the formation of amyloid polymers, which leads to a loss of its biological properties and limits its use as a potential therapeutic agent. Thus, a safe and effective method of stabilizing ovocystatin while maintaining its inhibitory activity was sought. Enzymatic dephosphorylation of the inhibitor, as well as an addition of trehalose or albumin, proved to help maintain the biological properties and activity of ovocystatin [33, 100].
While the studies on ovocystatin biological properties remain limited, the available data confirm the similarity to the well-evidenced cystatin C in several aspects, with special regard to antimicrobial, anticancer, and neuroprotective properties [107]. The antimicrobial effects of chicken cystatin were studied in relation to various microorganisms. Ovocystatin solution inhibits the growth of various Escherichia coli strains in vitro [108], while oral intake prevented symptoms of viral gastroenteritis in mouse models [109]. Similarly, ovocystatin combined with interferon-γ-(IFN-γ) can be used as a treatment for visceral leishmaniosis in mice, with its mechanism of action related to the increased production of nitric oxide in macrophages [110]. Studies on fungi revealed that ovocystatin inhibits the growth of Candida albicans with similar efficacy to fluconazole, whereas no fungal drug resistance to cystatin has been observed [111]. Interestingly, it has been also shown that ovocystatin induces actin cytoskeleton reorganization, as well as apoptosis, in cancer cells [107].
Due to the above-described characteristics of cystatin C and its similarity to ovocystatin, the influence of ovocystatin on cognitive functioning and neurodegeneration has also been explored. Previous research conducted with the use of ovocystatin in animal models revealed that its oral intake correlated with improved cognitive functions assessed with the Morris water maze. Interestingly, these effects were also observed in young rats [112]. Similar results were obtained in the animal model of B6C3-Tg transgenic mice used for the recreation of Alzheimer’s disease pathology. Six months of ovocystatin oral intake resulted in significantly improved spatial learning and memory of the experimental group. It has also been revealed that the survival of the mice during the 6-month study period suggests that ovocystatin might be safe to use. Nevertheless, toxicological studies are warranted to confirm this hypothesis [113]. In order to ascertain the underlying molecular mechanisms, a histopathological examination was conducted, revealing that both β-amyloid and tau protein deposits were significantly reduced in the hippocampi of the APP/PS1 mice treated intraperitoneally with ovocystatin [114]. With limited evidence for ovocystatin passage through the blood-brain barrier to the brain compartment, it is also possible that the observed effects were exerted peripherally and indirectly led to the changes in the brain tissue. In another study it was found that the use of ovocystatin in vitro led to reduced aggregation of 1-42 β-amyloid and in turn significantly decreased its toxicity in the PC12 cell line [115].
In conclusion, published studies present data on the endogenous role of cystatin C in the body. Few attempts have been made to administer exogenous cystatin C. Nagai et al. applied cystatin C in vivo directly into the hippocampus of rats, which led to a destruction of neurons in the dentate gyrus. However, immunohistochemical analysis showed there to be no amyloid-β formation after cystatin C administration [116].
Nutraceutical interventions have gained considerable attention in attenuating cognitive aging and its progression to dementia [117]. Ovocystatin, a cystatin isolated from chicken egg whites, is structurally similar to human cystatin C and its analogous form. Carrying out a series of preclinical studies to assess the effect of an exogenous administration of ovocystatin on cognitive impairment and Alzheimer’s disease seems justifiable with the potential for use as a procognitive and routinely used supplement to maintain cognitive function.