Oral squamous cell carcinoma (OSCC) is a highly malignant cancer of the oral cavity with poor prognosis. Currently, OSCC is treated with various surgical, radiotherapy, and chemotherapy methods. However, because of its specific anatomical location, surgical excision of OSCC tumour tissues is often technically challenging and leads to unavoidable injury to the surrounding anatomical structures [1].
Thus, various nanotechnologies are being explored as drug delivery systems (DDS) to address the shortcomings of chemotherapeutics [2]. DDS can either passively target tumours with increased cell membrane permeability and retention ability [3] or actively target cancer cells with decorated ligands that interact with or bind to receptors or proteins expressed on the surface of targeted cells [4]. The phosphorylated prodrug combretastatin A-4 phosphate (CA4P) is synthesised as a vascular disrupting agent (VDA). However, little is known about the anti-cancer and therapeutic activities of CA4P in OSCC.
Hyaluronic acid (HA) is an acidic polysaccharide containing D-glucuronic acid and N-acetyl-D-glucosamine, and has been explored as a drug carrier to deliver drugs to achieve targeted and synergistic combination therapy against cancers [5]. However, the anticancer activity of CA4P encapsulated in HA nanoparticles (NPs) in OSCC has not yet been extensively investigated.
Methods
HA-CA4P-NPs were prepared according to a previously reported method [6]. The size and zeta potential of the NPs were determined before subsequent experiments using a ZetaView® Nanoparticle Tracking Analyser with a helium-neon laser at a wavelength of 633 nm and a fixed scattering angle of 90°. The human OSCC cell line SCC-4 and human normal epithelial cells (HNECs) were cultured at 37°C in F-12 medium. CA4P released from the NPs was quantified using the dialysis method. Briefly, CA4P (1 mg) in lyophilised NP powder was dissolved in 5 ml of F-12 medium in a dialysis tube, which was then placed into a 200 ml falcon tube with 150 ml of F-12 medium and continuously stirred on a Cimarec stirring hot plate (Cole-Parmer, USA) at 37°C. The quantity of released CA4P was determined using HPLC, as previously reported [7].
To determine cellular delivery, HA-CA4P-NPs and CA4P were labelled with Texas Red fluorescent dye, added to F-12 medium containing cells at a final concentration equivalent to 50 µM CA4P, and cultured for an additional 24 h. The intensity of fluorescence emitted from the drugs was determined at different time points. To measure the cytotoxicity of NPs, cell viability was assessed using an MTT assay kit. Necrotic cells were detected using the YO-PRO-1/PI Apoptosis and Necrosis Assay Kit after the cells were inoculated with various concentrations of drugs and grown at 37°C in 5% CO2 for 24 h. The stained cells were loaded onto a FACSymphony Cell Analyser and analysed using the built-in software. To detect the levels of necrosis-related proteins, Western blot analysis was performed as previously described [8], using antibodies against Bax, RIP1, and RIP3. β-Actin was used as a loading control.
To assess pharmacokinetics, nude mice (BALB/c) were used. Animals were injected with CA4P and HA-CA4P-NPs (10 mg CA4P equivalent/kg) through the tail vein. At predetermined time points, venous blood was drawn to measure the CA4P content by HPLC. To assess antitumour activity, SCC-4 cells (106 cells/l) were subcutaneously injected into the right upper abdomen of nude mice as a xenograft mouse model. Six days after the injection, the animals were injected with 200 µl of PBS (control), CAP4, or HA-CAP4-NPs. Tumour development was monitored by measuring the longest axis of the mass.
Results
The mean size of HA-CA4P-NPs was ~85 nm, most NPs were between 55 and 130 nm, and the zeta potential was ~ –42 mV (Figure 1 A). HPLC analysis showed that the encapsulation efficiency of CA4P was ~84% in the NP preparation. CA4P was released from the HA-CA4P-NPs in a controlled and smooth manner. In the first 8 h, 15% of CA4P was released; the release slowed down after 16 h, and by 56 h, approximately 98% of CA4P was released from the NPs (Figure 1 B). In contrast, free CAP4 was released much faster and completed in less than 10 h (Figure 1 B). To investigate whether HA-CA4P-NPs selectively increased the delivery of CA4P into OSCC cells, fluorometry investigations showed that, compared with CA4P, HA-CA4P-NPs-incubated OSCC cells had nearly 10 times higher fluorescence intensity, indicating that HA-CA4P-NPs delivered more drug into the cells or bonded to the cell surfaces (Figure 1 C). Furthermore, SCC-4 emitted higher fluorescence than HNECs after incubation with HA-CA4P-NPs, but the fluorescence was low and similar between SCC-4 and HNECS after incubation with CA4P, indicating that HA-CA4P-NPs selectively targeted the cancer cells (Figures 1 C, D).
Figure 1
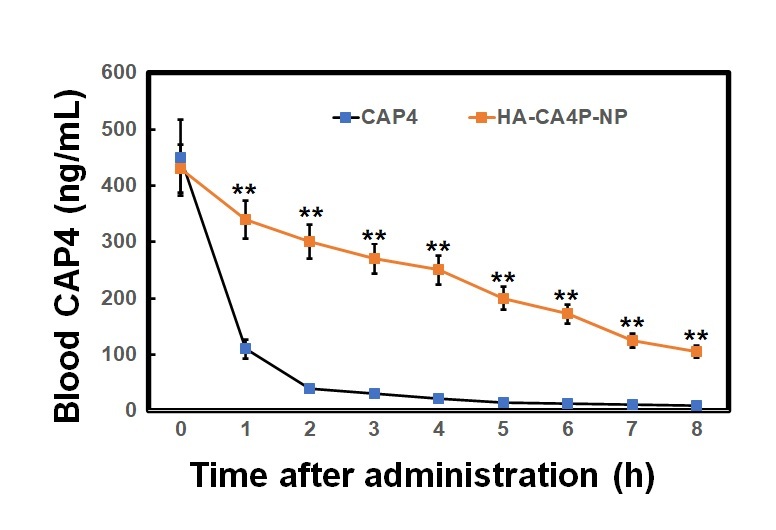
Preparation, releases, cellular cytotoxicity, drug dynamics and antitumour activity of HA-CA4P-NPs. A, B – The size distribution of HA-CA4P-NPs (A) and release profile of HA-CA4P-NPs (B). HA-CA4P-NPs and CA4P were added to F-12 medium in a dialysis tube and gently stirred in F-12 medium at 37°C. Samples were taken at different times to quantify released CA4P using HPLC. C, D – Cellular delivery of CA4P and HA-CA4P-NPs in HNECs and SCC-4 cells. HNECs and SCC-4 cells were incubated with 50 μM fluorescent dye Texas Red labelled drugs for 24 h. The cells were then harvested by centrifugation at 500 g and room temperature for 10 min, rinsed 5 times with pre-chilled PBS (pH7.4) buffer, loaded onto a AquaMate 7100 fluorometer (Themo Scientific, USA) to determine the intensity of fluorescence emitted from the drugs at 560 nm after excited at 488 nm. C – Fluorescence intensity after incubation with CA4P and HA-CA4P-NPs. D – representative microscopy photos of CA4P and HACA4P-NPs-treated HNECs and SCC-4 cells. DAPI was used to stain the nucleus. E–G – Cell viability, necrosis and protein expression after exposed to CA4P and HA-CA4P-NP. HNECs and SCC-4 cells were treated with 0 to 40 μM CA4P and HA-CA4P-NP for 48 h. The viability and necrosis of cells were determined using MTT assays and flow cytometer (E, F), respectively. Expression of necrosis-related proteins was analysed using Western blots (F). F – left panel, flow cytometry results of necrosis assay; right panel, statistical analysis of necrotic cell. **, ##p < 0.01 vs. CA4P and HNECs, respectively. *,**p < 0.05 or < 0.01 vs. 0 μM HA-CA4P-NP. G – left panel, representative Western blots; right panel, statistical analysis of protein levels. H – Dynamics of serum CA4P levels in mice injected with CA4P and HA-CA4P-NPs. Mice were injected with CA4P and HA-CA4P-NPs at 10 mg/kg and blood samples were taken to measure serum CA4P levels at different time points using HPLC. **p < 0.01 vs. CA4P. I, J – Tumour growth in xenograft models of mouse after treatment with HA-CA4P-NPs and CA4P. Mice were injected with SCC-4 cells to establish xenograft models. The tumour-bearing mice were treated with CA4P and HA-CA4P-NPs. I – Tumour size at different days after drug treatment. J – Isolated tumours at final day of drug treatment. D **p < 0.01 CA4P
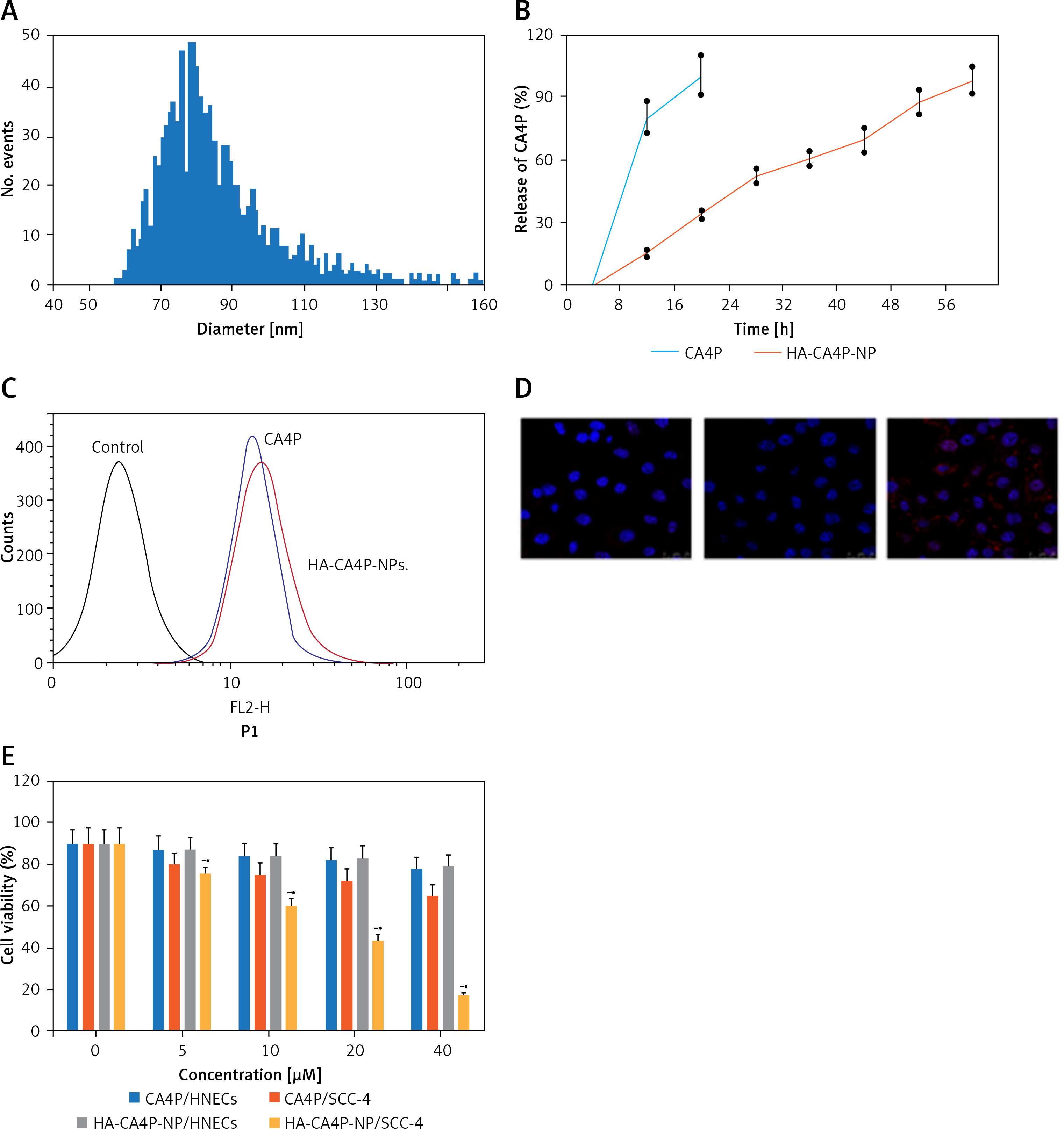
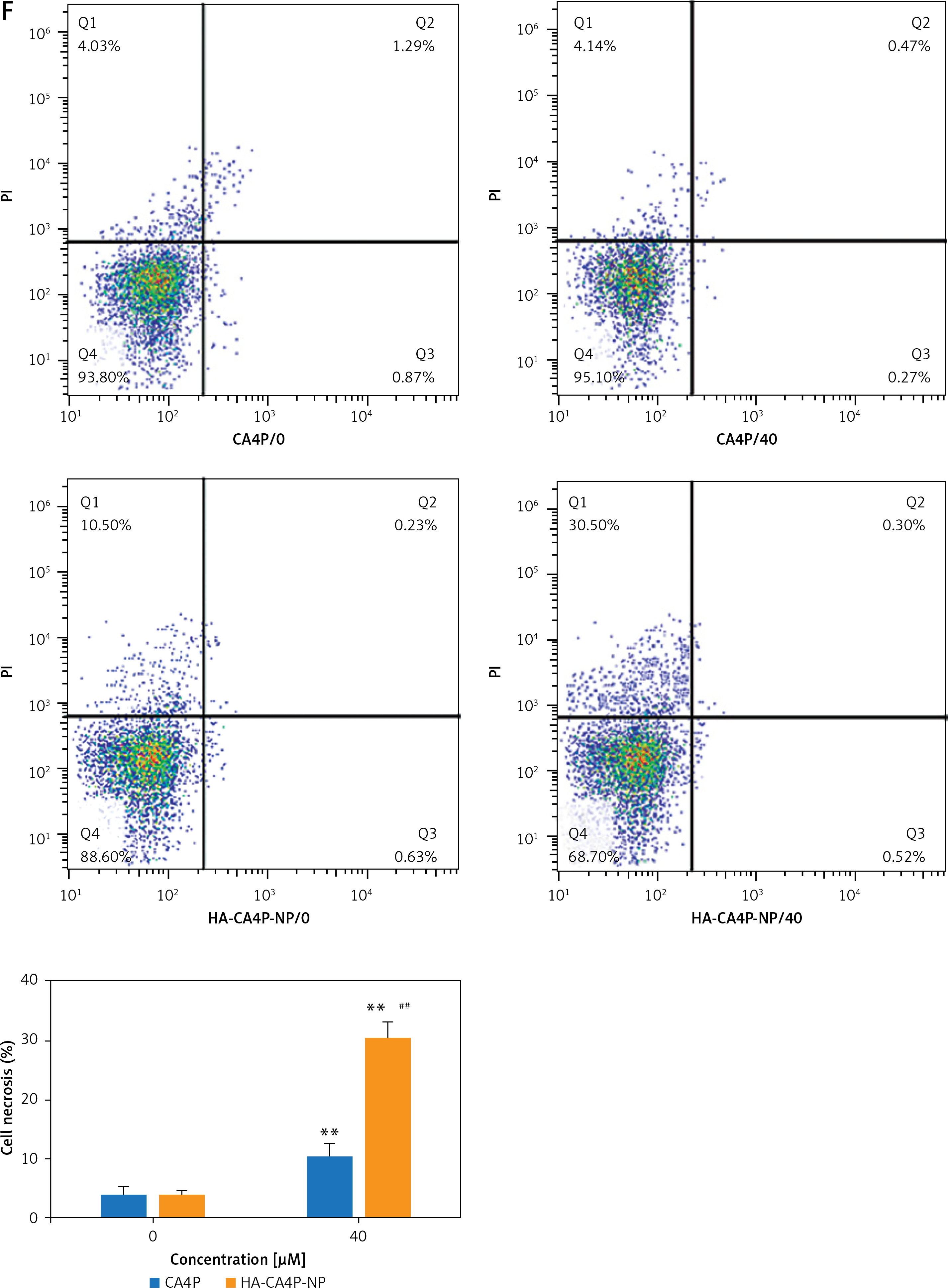
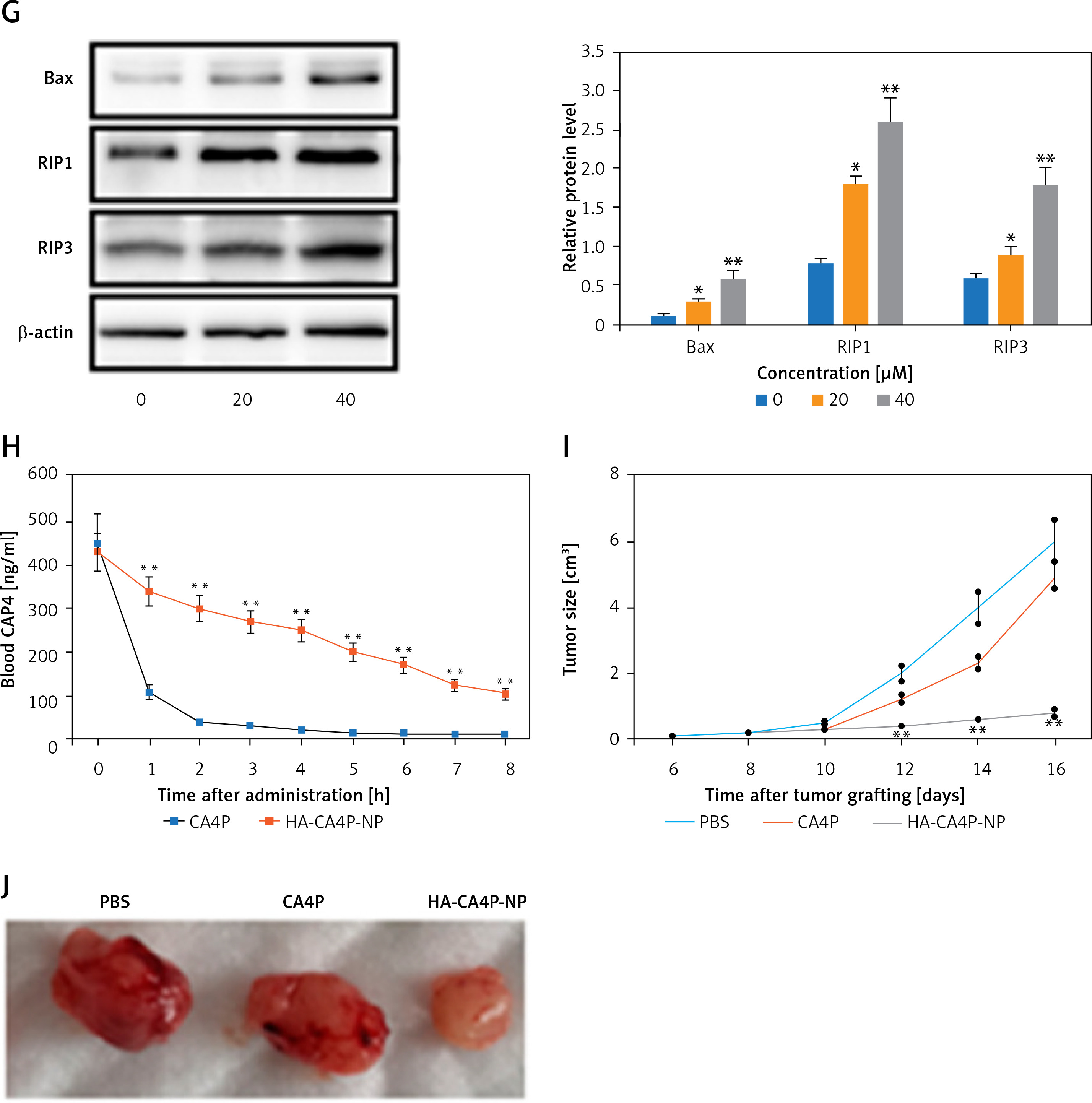
Cytotoxicity was compared using the HNECs and SCC-4 cells. The viability of SCC-4 cells after exposed to 40 µM HA-CA4P-NPs and CA4P decreased from 90% at 0 µM to 17% and 65% at 40 µM (Figure 1 E). In contrast, both HA-CA4P-NPs and CA4P had low toxicity to HNECs in the same concentration range as SCC-4 cells (Figure 1 E). Furthermore, flow cytometry showed that after incubation with the drugs at 40 µM for 48 h, HA-CA4P-NP led to 30% necrotic SCC-4 cells, which was 3 times after CA4P treatment (Figure 1 F). Western blot analysis showed that NP-treated OSCC cells had significantly higher expression of necrosis-related proteins Bax, RIP1, and RIP3, suggesting that programmed necrosis was activated, particularly after NP treatment (Figure 1 G).
To assess drug pharmacokinetics, HA-CA4P-NPs and CA4P were administered to mice and CA4P circulation was assessed. The data revealed that free CA4P was cleared very quickly in the blood, and its concentration declined remarkably in 2 h and was barely detected at 5 h after administration. However, for the HA-encapsulated CA4P, the circulation time was significantly prolonged. Six hours later, the blood CA4P concentration was still very high in HA-CA4P-NP-administered mice (Figure 1 H). Compared with free CA4P, the area under the curve (AUC) and mean residence time (MRT) of HA-CA4P-NPs were more than 3 times that of free CA4P, and the elimination rate constant ke was similar (Table I). The antitumour activity of HA-CA4P-NPs was assessed using xenograft models. The results showed that the tumours grew slowly in the first 10 days after the injection of SCC-4 cells and quickly in PBS- and CAP4-treated mice; however, growth was strongly suppressed in HA-CA4P-NPs-treated mice (Figures 1 I, J). Sixteen days after grating, the weights of the tumours in the HA-CA4P-NPs-treated mice were only one-fifth of those in the CA4P-treated mice (Figures 1 H, J).
Discussion
In this study, we prepared HA-CA4P-NPs with a mean size of 85 nm and analysed their potential for OSCC treatment. Drug release and delivery studies showed that CA4P was released from HA-CA4P-NPs in a controlled and smooth manner. HA-CA4P-NPs generated more fluorescence than CA4P in SCC-4 cells and had higher toxicity to SCC-4, but not to HNECs. Pharmacokinetic analysis showed that HA-CA4P-NPs had better drug kinetics, and animal experiments demonstrated that the NPs had significantly stronger antitumour activity than CAP4. These findings indicate that HA-CA4P-NP is a potent drug delivery system for OSCC and should be further explored for its therapeutic efficacy in OSCC and other cancers.
To address the issues associated with chemotherapy, various nanotechnology-based drug carrier systems have been explored to improve the selectivity and delivery efficacy of the drug target with a higher drug concentration in the target lesion for potential applications in cancers, including OSCC [9, 10]. Polymeric nanoparticles, nanoliposomes, solid lipid nanoparticles, and receptor-mediated drug delivery systems have been studied to reduce systemic toxicity, nephrotoxicity, neurotoxicity, and gastrointestinal toxicity; improve therapeutic effects; and prevent drug resistance [11, 12]. The mean size of HA-CA4P-NPs was 85 nm, which is in the desired range reported to avoid rapid kidney clearance [13]. The in vitro release of HA-CA4P-NPs was much slower than that of free CA4, suggesting that HA-CA4P-NPs functioned as a slow-release system for CA4. At the same time, HA-CA4P-NPs targeted OSCC more than free CA4P did. HA has high affinity for CD receptors, which are highly expressed on the surface of cancer cells. This might explain the selective delivery of HA-CA4P-NP to OSCC cells. Low fluorescence was generated after incubation of HA-CA4P-NP with non-cancer HNEC, further indicating that HA-based NPs actively target cancer cells, probably because RHAMM receptors are highly expressed in cancer cells [14].
We found that the toxicity of CA4P in OSCC cells was low. However, high cytotoxicity was observed when HA-CA4P-NP were used to treat OSCC cells, although its toxicity against HNECs was low. This was probably due to increased delivery of the drug to OSCC cells. The low toxicity of HA-CA4P-NP to HNECs suggests that it has excellent biocompatibility with non-cancer cells and is suitable for systemic administration in cancer treatment. As a tubulin-depolymerising agent, CA4P induces extensive ischaemic necrosis in various cancers [15]. Flow cytometry analysis revealed that SCC-4 cells had a high percentage of necrotic cells after HA-CA4P-NP treatment, and proteins related to necrosis were upregulated, indicating that HA-CA4P-NPs probably activated the ROS/JNK/c-Jun and RIP1/RIP3/MLKL pathways [16].
Improved pharmacokinetics were observed in mice treated with HA-CA4P-NPs compared with those treated with CA4P. This was likely due to the higher stability and controlled release of CA4P encapsulated in the HA. Previously, the encapsulation of drugs in liposome-derived NPs was shown to slow in vivo clearance [17]. Therapeutic efficacy studies showed that compared with CA4P, HA-CA4P-NPs had much stronger antitumour activity, which is probably due to the improved pharmacokinetics and targeted delivery of HA-CA4P-NPs to cancer cells.