Introduction
Precocious puberty is defined as the occurrence of secondary sex characteristics under 8 years of age in girls and under 9 years of age in boys. Depending on the etiology, precocious puberty can be divided into two main groups. The first one includes central (gonadotropin-releasing hormone [GnRH]-dependent) precocious puberty (CPP), which results from premature activation of the hypothalamic–pituitary–gonadal axis. The second group includes peripheral (pseudopubertal, GnRH-independent) precocious puberty (PPP) in which sex steroid secretion is independent of the GnRH pulse generator in the hypothalamus. The aim of this paper is to discuss current data on the causes of CPP with particular attention given to neurological aspects.
Methods
The key criteria of literature reports’ assortment was to find medical data focusing on the neurological causes of premature puberty. We reviewed literature from 1995 to 2019 using PubMed. Studies with a limited number of patients and studies not exactly correlated with neurological disorders were not included. Additionally we analyzed 3 articles, because they are the only works describing cases of CPP as a complication of tuberculous meningitis (from the years 1959 and 1970), and one describing puberty in the septo-optic dysplasia syndrome (1989).
Epidemiology
There are very few studies analyzing the prevalence and etiopathogenesis of CPP. It has been confirmed that this rare disorder is much more prevalent among girls because the activation of the hypothalamic–pituitary–gonadal axis is related to a weaker stimulus needed for GnRH stimulation in females [1]. Based on a Korean study conducted between 2008 and 2014, it was concluded that the overall incidence of CPP during the study period was 122/100,000 persons (i.e. girls 262/100,000; boys 7/100,000) [2].
Pathomechanism
The exact mechanism controlling the onset of puberty is still not fully understood. It is confirmed that gonadotropins (follicle-stimulating hormone [FSH] and luteinizing hormone [LH]) are secreted in a pulsatile manner from the anterior pituitary lobe into the systemic circulation as a result of episodic and pulse secretion of GnRH by hypothalamic cells (mainly of the arcuate nucleus). By acting on the target cells in the gonads, gonadotropins lead to an increase in the production of sex steroids, which, in turn, are responsible for the development of secondary sex characteristics. The GnRH pulse generator is influenced by a number of stimulating and inhibiting factors, including neurotransmitters, environmental factors and even the nutritional status.
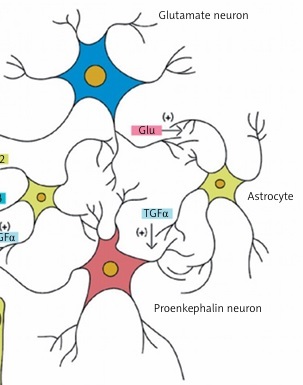
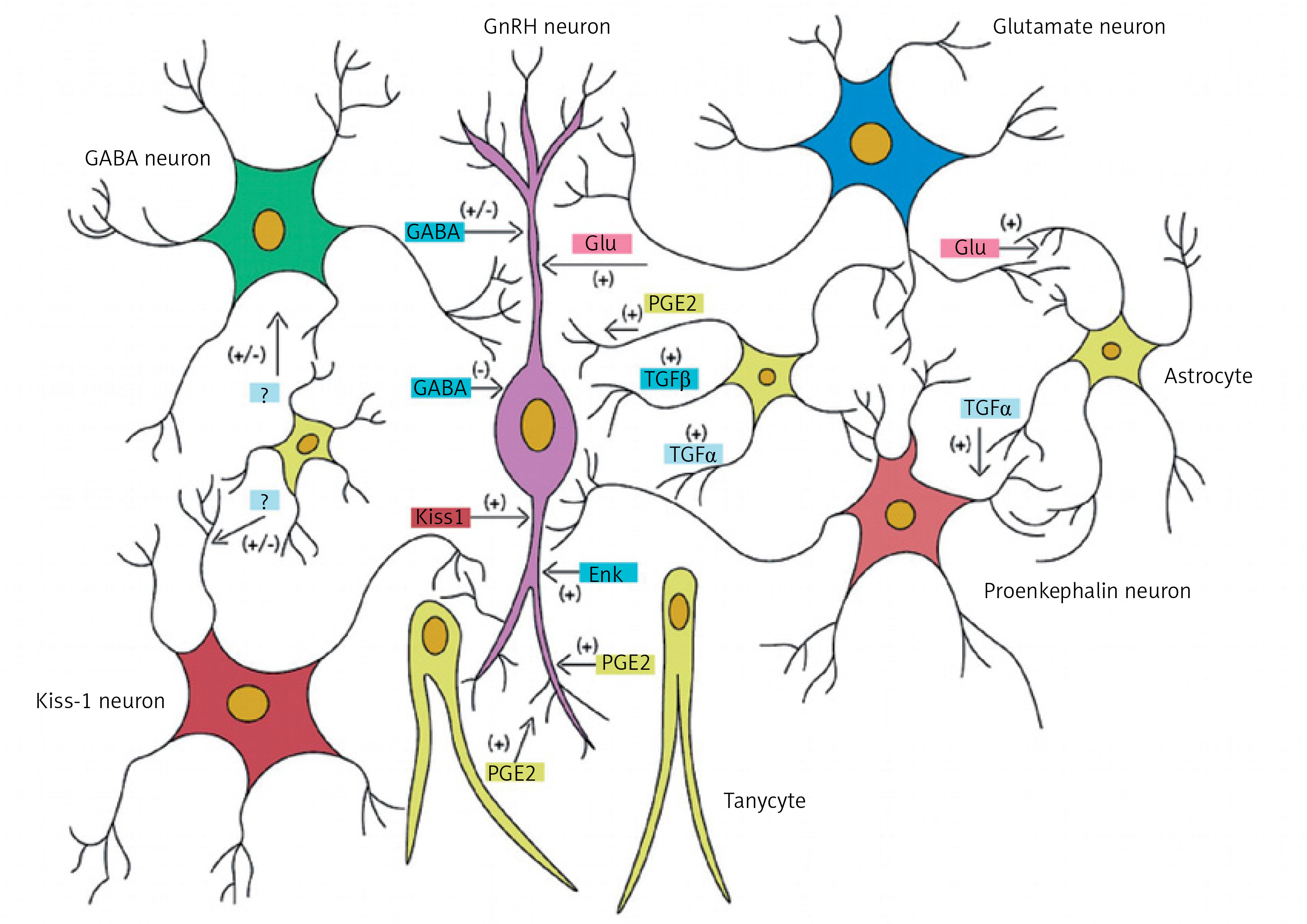
Ojeda et al. reported that the central control of puberty is mainly dependent on the expression of genes encoding both proteins stimulating GnRH release (e.g. glutamate, kisspeptin) and proteins inhibiting GnRH release (e.g. γ-aminobutyric acid [GABA], opioids) [3]. Figure 1 shows the process of influence of neurotransmitters and other factors on GnRH-secreting neurons. Kisspeptin plays a major role among the stimulating factors. It is influenced by neurokinin B and dynorphin. These three regulators compose the kisspeptin-neurokinin B-dynorphin neuronal (KNDy) system, a key player in pubertal onset and progression) [4]. However, in childhood, the suppression of GnRH secretion seems to be regulated mainly by GABA. Damage to the central nervous system (CNS) due to the increased intracranial pressure or tumor may eliminate this inhibitory effect and lead to precocious puberty [1].
Figure 1
Cellular and molecular mechanisms contributing to GnRH release by normal human hypothalamus. Excitatory neurons (producing kisspeptin and glutamatergic) and inhibitory (GABAergic) neurons interact directly on the GnRH neuron in the hypothalamus. Some GnRH neurons show a paradoxical stimulation by GABA. Glial cells also have a stimulatory effect mediated by the transforming growth factor α (TGFα), prostaglandin E2 (PGE2) and other factors (own data modification based on Lomniczi A, Matagne V, Ojeda SR, Neuroendocrinology of puberty. In: Squire LR (ed). Encyclopedia of Neuroscience. Elsevier, London, 2009)
Diagnostic assessment
A GnRH test (synthetic gonadoliberin or its analog) is the gold standard. LH increase by 3–4 U/l and FSH increase by 2–3 U/l above baseline concentrations are observed in healthy children in prepubertal age after GnRH stimulation [1]. Currently, it is known that an increase in LH ≥ 5 IU/l [5] and the LH/FSH ratio > 0.66 after GnRH analog stimulation allow the diagnosis of CPP [6].
Complications
Untreated CPP leads to accelerated bone age and premature epiphyseal closure, which ultimately leads to reduced final height. Early occurrence of pubertal characteristics and high concentration of hormones may contribute to behavioral disorders – physical maturity does not correspond to mental maturity. In addition, early menarche in girls is a risk factor for the development of ischemic heart disease [7], breast cancer [8] and endometrial cancer in adulthood [6].
Treatment
Pharmacotherapy is based on long-acting GnRH analogs which as a result of permanent exposure of the GnRH receptor to its ligand lead to inhibition of the hypothalamic–pituitary–gonadal axis. They are synthetic preparations mimicking the structure of the native GnRH decapeptide with D-tryptophan substituted for the naturally occurring L-tryptophan, which significantly prolongs receptor excitation. This results in stimulation (mostly after the first two doses) with subsequent inhibition of receptor activity and downregulation, which leads to inhibition of gonadotropin secretion. Lack of receptor response is due to the separation of the receptor from the intracellular effector pathway of the transmitted stimulus.
Treatment with GnRH analogs is safe and well tolerated [9]. Preliminary treatment assessment shows a positive effect on overall growth. However, treated patients do not reach the final height that results from the average growth of their parents. It is due to the onset of the pubertal growth spurt, acceleration of bone age with inadequate child growth and no further growth after the discontinuation of pharmacologic treatment (the growth spurt is slowly inhibited during GnRH analog treatment) [1].
Etiology
The etiology of CPP is very diverse. In girls, idiopathic CPP accounts for the vast majority of cases. However, recently the incidence of idiopathic cases has decreased due to the detection of gene mutations that lead to premature activation of the GnRH pulse generator. In boys, however, organic changes in the CNS are much more prevalent (40–75% of cases), which prompts imaging assessment of the CNS in each case of CPP among boys. In the female sex with CPP, organic CNS changes on MRI are reported in 0–27% of cases. However, they are more frequently detected in girls in whom secondary sexual characteristics occurred very early, i.e. below 6 years of age [10]. Young age, rapid progression, and high estradiol concentrations are the factors that might predict increased risk of brain abnormalities [11].
Table I presents the causes of CPP with the division into those due to congenital structural changes in the CNS, acquired changes and those unrelated to the pathology in the CNS. Table II presents MRI results of a group of women with CPP – based on Orsdemir-Cantas et al. [10].
Table I
Causes of central precocious puberty
Table II
Magnetic resonance imaging pathological findings of a group of women with central precocious puberty, N = 142 (based on [10])
Central precocious puberty due to congenital structural changes in the central nervous system
Hypothalamic hamartomas
Hypothalamic hamartomas (HHs) are the most common organic changes in the CNS associated with CPP. These are benign, congenital tumors, composed of ectopic neurosecretory neurons and bundles of nerve fibers and glia. The prevalence of HH is 1/50,000–1/100,000 children. Magnetic resonance imaging shows a characteristic image of the mass mostly adjacent to the posterior part of the hypothalamus located between the tuber cinereum and mamillary bodies. In a study on 277 patients with HH based on CNS imaging, 63% developed CPP symptoms and 61% developed symptoms of epilepsy [12]. Those patients were asymptomatic or presented with mental disability. Clinical symptoms are closely related to the location of the tumor. Extrahypothalamic and pedunculated tumors are usually associated with CPP, whereas intrahypothalamic non-pedunculated tumors are connected with epilepsy. Hypothalamic hamartoma occurs in both CPP and epilepsy in 27% of cases [12].
The pathomechanism of CPP in HH patients is not well understood. Currently, several hypotheses are considered: a) HH acts as an ectopic GnRH pulse generator; b) HH is composed of transforming growth factor α (TGF-α) producing astroglial cells that could cause premature activation of pulsatile GnRH release; c) due to increased CNS pressure associated with the presence of tumor mass, reduced influence of inhibitory neurotransmitters on GnRH neurons in the hypothalamus is observed [11, 13].
Symptoms of precocious puberty, in this case, occur very early, mostly below 4 years of age. The median of secondary sex characteristics in girls is 6 months (0.4–7 years) whereas in boys it is 2 years (0.5–5 years) [14]. Epileptic seizures in the form of characteristic laughing or crying occur mainly in childhood. Later they are diagnosed as typical generalized or focal seizures. In patients with epilepsy, delayed psychomotor development is observed, which may be secondary to seizures (due to epileptic encephalopathy). Moreover, the level of mental disability correlates with the frequency and severity of seizures. Patients who present only with CPP symptoms without epilepsy show a normal intelligence quotient. Neurosurgical treatment of HH is reserved for cases with refractory epilepsy.
Arachnoid cysts
Arachnoid cysts are relatively rare intracranial lesions, which are mostly congenital. However, they may also occur as a result of infection, trauma and CNS bleeding. They extend from the arachnoid mater, which (by splitting or duplication) forms a space with fluid that is similar to the cerebrospinal fluid. The middle and posterior cranial fossa is the most common location of arachnoid cysts. Such cysts are characterized by significant dynamics of change. They can grow, decrease in size or disappear spontaneously. Their presence results in neurological and ophthalmological complications in 30–40% of cases and endocrine complications, of which CPP and growth hormone (GH) deficiency are the most prevalent [15].
Mohn et al. described 6 cases of children with arachnoid cysts [15]. Most patients presented with neurological symptoms such as headaches, intracranial hypertension, episodes of apnea, generalized epileptic seizures, macrocephaly, delayed psychomotor development and ophthalmological complaints. Endocrine complications included CPP (present in 3 of these 6 patients), GH deficiency (present in 4 out of 6 patients), hypopituitarism due to TSH and ACTH deficiency (in 2 out of 6), diabetes insipidus (in 2 out of 6) and hypogonadotropic hypogonadism (in 1 out of 6). It is suspected that premature activation of the GnRH pulse generator is a response to the cyst mass effect on the hypothalamic region.
Myelomeningocele, hydrocephalus, Chiari malformation
Increased intracranial pressure in the prenatal period, causing dilatation of the third ventricle, and early symptoms of brain stem dysfunction are significant risk factors for early and precocious puberty [16, 17]. The incidence of myelomeningocele (MMC) was 3.8/10,000 births in the 1990s. Currently, the incidence is constantly decreasing, mainly due to an increase in the number of abortions of fetuses with congenital malformations. Hydrocephalus develops in more than 80% of children with MMC, most of whom undergo ventriculoperitoneal shunt implantation to decrease intracranial pressure. In addition to typical problems, such as lower limb paresis, neurogenic bladder dysfunction, orthopedic abnormalities and intestinal complaints, symptoms of early and precocious puberty develop in about 52% of girls and 21% of boys with MMC. Sex-related differences are mainly due to the fact that the male sex is generally less predisposed to the development of CPP [16].
The Arnold-Chiari syndrome is a congenital cerebral malformation in which the rhombencephalic structures, depending on the subtype of malformation, are displaced into the spinal canal. It may coexist with MMC and hydrocephalus or it can be an isolated defect. While Chiari type II malformation causing hydrocephalus and an increase in intracranial pressure seems to be clearly related to the development of CPP, the cause of the increased risk of CPP remains unknown in the case of Chiari type I malformation (where hydrocephalus does not occur) [16].
Septo-optic dysplasia
Septo-optic dysplasia (SOD; de Morsier syndrome) is a rare, genetically conditioned disease. The disorder is characterized by underdevelopment of the optic nerve disc, midline malformation of the brain, including the absence or underdevelopment of the transparent septum and the corpus callosum, and hypopituitarism diagnosed at different age. The etiology remains unknown. The relationship with mutations in the HESX1 homeotic gene was observed in rare cases of familial history of the syndrome [18].
Delayed puberty is much more prevalent in patients with SOD. However, cases of co-occurrence of SOD and CPP have been reported in the literature. Hanna et al. observed the occurrence of early and precocious puberty in 3 of 13 patients with SOD [19]. Ladjouze et al. reported CPP in 4 patients in whom SOD and GH deficiency had previously been diagnosed [20]. Relatively rare co-occurrence of somatotropin deficiency and the development of CPP is more prevalent in patients with SOD in whom an arachnoid cyst was also noted [21]. Although the mechanism of CPP development in patients with SOD remains unknown, it is presumed to be due to structural changes in the hypothalamus.
Therefore patients with bilateral optic nerve hypoplasia should be examined for anterior pituitary hormone deficiency. However, inadequate acceleration of the growth rate and bone age may be predictive for CPP development during the follow-up of patients with SOD treated with growth hormone due to the coexisting somatotropin deficiency.
Central precocious puberty due to acquired structural changes in the central nervous system
Tumors
Tumors of the CNS are the second most common cause of childhood cancer. However, they rarely cause CPP. Symptoms of tumors of the CNS depend on their size, location and the age of the child. In patients with CPP, MRI images reveal the following types of tumors: astrocytomas, optic nerve gliomas, craniopharyngiomas, hypothalamic tumors and tumors of the pons, suprasellar tumors, fourth ventricle tumors, pineal tumors, neuroblastoma (including neuroblastoma with cerebral metastases), germ-cell tumors, retinoblastoma, pituitary macroadenoma, Ewing’s sarcoma, Burkitt’s lymphoma and Langerhans cell histiocytosis [10, 22].
Of the above examples, pilocytic astrocytoma is the most common cause of CPP. The exact frequency of pilocytic astrocytoma in children with CPP is not established yet (according to our knowledge). Astrocytomas were found in 15 out of 142 MRI pathological findings in the study population with CPP of Cantas-Orsdemir et al. This result classifies them in the first place among brain tumors [10]. Astrocytoma is a tumor with low malignancy and a rare malignant transformation. The peak incidence rate is 3–7 years of age. It often occurs in patients with type 1 neurofibromatosis. In such cases its location in the optic chiasm and hypothalamus significantly increases the risk of CPP [23].
Previous radiotherapy
The prevalence of CPP increases following cranial exposure to radiation due to tumors or leukemia. Low doses of radiation (18–24 Gy) used in the prophylactic treatment of the CNS in acute lymphoblastic leukemia and moderate doses (25.0–47.5 Gy) used in the treatment of brain tumors in children increase the risk of CPP [1]. Radiation therapy can destroy GABA-secreting neurons, leading to decreased GABA concentration and a loss of the inhibitory effect on the GnRH pulse generator. Additional risk factors for CPP include the female sex and the young age of the child undergoing treatment. Of note, the clinical picture of CPP may be altered due to the coexistence of GH deficiency in children who underwent radiotherapy. Higher doses of radiation usually result in gonadotropin deficiency [24].
Traumatic brain injury
A prospective Czech study on hypothalamic– hypophyseal dysfunction in children and adolescents following traumatic brain injury showed that insufficiency of the hypothalamic–hypophyseal axis was more prevalent than the premature activation of the axis with respect to gonadotropin secretion. Of 58 patients, 23 had a Glasgow Coma Scale score < 8, which corresponded to severe brain damage. Hypogonadotropic hypogonadism occurred in 25% of the teenagers, whereas CPP symptoms developed in 2 boys [25].
Of note, however, the median age at the time of injury was 11.3 years, which means that most children were already pubertal at the time of injury (Tanner ≥ II). The risk factors for the development of the dysfunction of the hypothalamic–hypophyseal axis include the gravity and the extent of the injury on MRI.
In another prospective study conducted by Dassa et al. on 61 children after acute brain injury, 18 children developed GH deficiency, and 4 children central precocious puberty 5.7 years after injury. No hypogonadotropic hypogonadism was observed. Severe traumatic brain injury in childhood can lead to permanent pituitary dysfunction; GH deficiency and CPP may appear after many years [26].
Systematic hormonal assessment in children after severe traumatic brain injury and prolonged monitoring of growth and pubertal maturation should be performed in every case.
Neuroinfections
So far, few cases of CPP have been reported. These cases were the complication of past meningitis and encephalitis. However, there were single reports published in the 1950s and 1960s which showed such a relationship, especially after tuberculous meningitis [27, 28].
Currently, no such data are available, which is probably due to the fact that severe forms of tuberculosis (including tuberculous meningitis) are extremely rare at present.
Perinatal hypoxic-ischemic encephalopathy
Perinatal hypoxic-ischemic encephalopathy with cerebral palsy (which is a complication of encephalopathy) is another risk factor for CPP [23]. This is probably due to an early brain development disorder secondary to hypoxia. However, the pathomechanism remains unknown.
Metabolic diseases
These are rare genetically determined diseases in which there is an absence or deficiency of the enzyme involved in the metabolic pathway. As a result, the accumulation of a substrate and the deficit of the product are observed. Sometimes an alternative pathway is established. Symptoms of CPP can potentially occur in any metabolic disorder in which there is accumulation of excess undissolved substrates in the white matter of the brain. So far, single cases of lysosomal storage diseases have been described. Beside multiple neurological symptoms, CPP also developed in these disorders. These include Tay-Sachs disease [29], metachromatic leukodystrophy [30] and ceroid lipofuscinosis [31]. The authors of those reports suggested that the material accumulated in the white matter adversely affected the GnRH pulse generator, damaging its inhibitory pathways.
Central precocious puberty not related to central nervous system structural changes
Idiopathic precocious puberty
The cause of CPP remains undetected among girls in more than 90% of cases (idiopathic precocious puberty). Idiopathic precocious puberty is significantly less prevalent among boys and CPP is mostly the result of lesions in the CNS [1]. Of note, the number of cases diagnosed with the idiopathic form of the disease is steadily decreasing, which is mainly related to the detection of mutations that cause CPP [9]. However, genetic testing for these mutations is still rarely performed.
Genetic factors
First reports on the occurrence of genetic causes of CPP date back to 2004. De Vries et al. observed that the incidence of familial CPP was 27.5% in their study group [32]. Over time, mutations in various genes were detected, which resulted in the occurrence of CPP (gain-of-function mutations in the genes encoding kisspeptin [KISS1], kisspeptin receptor [KISS1R] and tachykinin 3 [TAC3]; loss-of-function mutation in genes encoding makorin ring finger protein 3 [MKRN3]) [11, 33].
MKRN3 gene defects are a frequent cause of familial CPP [11]. This disorder has been associated with normal MRI scans of the hypothalamic-pituitary region. It is currently considered that genetic analysis of this gene should be included in the routine clinical investigation of familial and idiopathic cases of CPP.
In addition, chromosomal disorders (mainly deletions) are also involved in many genetic syndromes associated with a higher risk of CPP. These abnormalities include 1p, 7q (Williams-Beuren syndrome), 9p, 14q, 15q (Prader-Willi syndrome) and Xp 22 (Rett syndrome).
Environmental factors
Adopted children are at greater risk of early or precocious puberty. The exact mechanism explaining the relationship between adoption and CPP remains unknown. Current hypotheses consider the influence of race, emotional, nutritional and environmental factors [34]. Different stress situations (such as adverse events, intensive physical training, psychological conditions and illnesses) are known to suppress the hypothalamic–pituitary–gonadal axis. In this context, it is possible that in foreign migrating children, withdrawal from a stressful environment contributes to potentiation of maturation [4].
In addition, there are reports on adverse effects of pesticides on the endocrine system. A French epidemiological study conducted in the regions where the use of pesticides was common showed a significantly higher prevalence of CPP in children compared to those who lived in less polluted areas [35]. DDT [1,1-dichloro-2,2-bis (4-chlorophenyl) ethane] and some isomers behave as estrogen agonists or may promote the hypothalamic–pituitary-gonadal axis maturation [4].
Symptoms of early and precocious puberty are more prevalent among obese girls, which is probably related to an increase in leptin levels. This relationship is not so strongly visible among the male sex. Leptin is produced in fat cells and suppresses appetite after receptor binding in the hypothalamus. It plays a major role in the maturation of mice and rats. However, in the case of leptin deficiency, delayed puberty was observed in humans and an increase in gonadotropin levels was reported following leptin administration. These data indicate that leptin may act as a trigger for the initiation of puberty. However, clinical trials are inconclusive [1].
Impact of antiepileptic drugs
There are several studies suggesting that antiepileptic drugs, especially valproic acid, may contribute to the development of CPP. Many patients with CPP symptoms are also affected by epilepsy. However, clear evidence to support this theory has not been found yet [36].
Impact of long-term exposure to sex steroids
Long-term exposure to sex steroids (in the case of congenital adrenal hyperplasia, testotoxicosis and sex steroid-producing tumors) can result in secondary CPP. Sex steroids can lead to maturation of CNS centers. The decrease in these hormones during treatment of the primary underlying disorder causes activation of the precociously matured hypothalamic GnRH pulse generator via feedback mechanisms [11].
Conclusions
Central precocious puberty is a relatively rare disease. However, currently, there are too few studies on the prevalence of this disorder, the pathomechanism and the causes of CPP among children. It is confirmed that it accompanies many pathologies of the CNS and frequently coexists with neurological symptoms resulting from these abnormalities. Structural changes in the CNS associated with CPP are: hypothalamic hamartoma, arachnoid cyst, myelomeningocele, hydrocephalus, Chari malformation, septo-optic dysplasia and tumors. Furthermore, cranial exposure to radiation, traumatic brain injury, neuroinfections, perinatal hypoxic-ischemic encephalopathy and some metabolic diseases are also risk factors for CPP. Safe and effective treatment is available. The time of treatment implementation is crucial to the successful outcome. Further studies are warranted to better analyze and understand the pathomechanism of CPP, including the identification of real risk factors for CPP in children.