Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an uncommon arrhythmogenic disorder characterized by adrenergic-induced bidirectional and polymorphic VT occurring in young patients with a structurally normal heart [1–3]. Although the actual prevalence of CPVT is unknown, it is estimated at 1 : 10,000 [4]. We present the case of a 12-year-old girl suffering multiple falls while cycling finally diagnosed with CPVT and found to be heterozygous for a pathogenic RYR2 gene mutation. She did not have a family history of syncope or sudden cardiac death and was referred because of suffering multiple falls and minor trauma while riding her bicycle starting at age 8 years. Initially, her parents attributed the falls to inexperience but sought medical assistance when tonic movements of both arms occurred during one of the falls. The first clinical diagnosis was epilepsy, and, without further studies, she was treated with magnesium valproate for 6 months without clinical improvement. Due to the lack of response and the correlation of falls with exercise she was referred for evaluation to the Electrophysiology Department of the Hospital General Naval de Alta Especialidad.
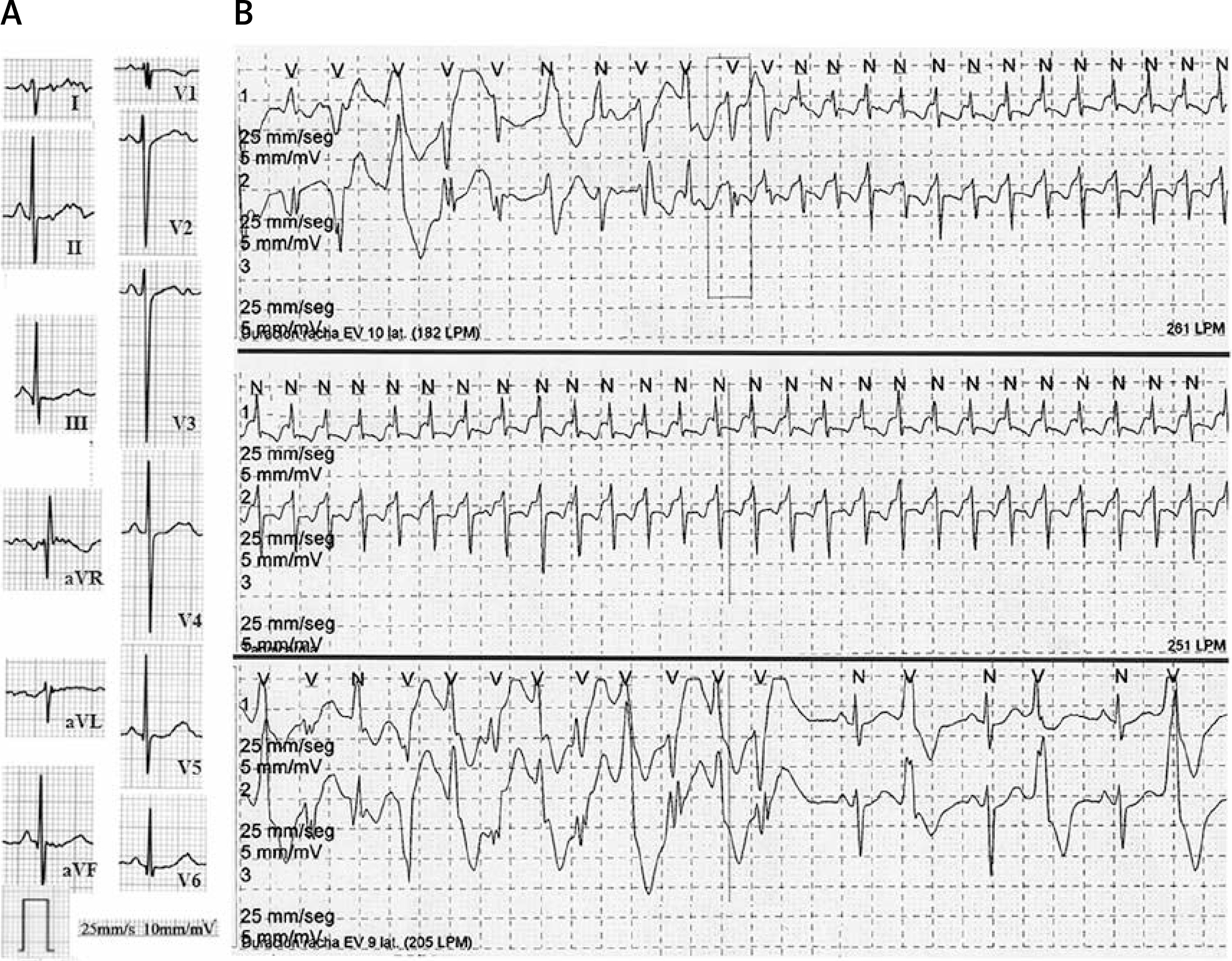
On interrogation, the child (and her mother corroborated) had reduced exercise tolerance, quick fatigue, pallor, shortness of breath, and fast and irregular heartbeats associated with exercise. Physical examination was normal. The ECG showed normal P wave, PR, and QT intervals; only a fragmented QRS complex and nonspecific T wave abnormalities were observed, particularly in precordial leads, where a small notch in the descending limb of the T wave could be found (Figure 1 A). The chest X-ray and echocardiogram were normal. Holter monitoring (Figure 1 B) showed frequent ventricular premature beats (VPB) with multiple morphologies (14%, n = 1603), and presence of couplets and triplets (pleomorphic), reproducible with heart rates above 110 bpm. Also, a self-limited supraventricular tachycardia at 261 bpm was documented.
Figure 1
A – 12-lead ECG (25 mm/s, 10 mm/mV). B – Holter monitoring. The first strip shows the beginning of the non-sustained narrow QRS tachycardia that continues in the second strip. On the third strip, bidirectional ventricular tachycardia is followed by bigeminy
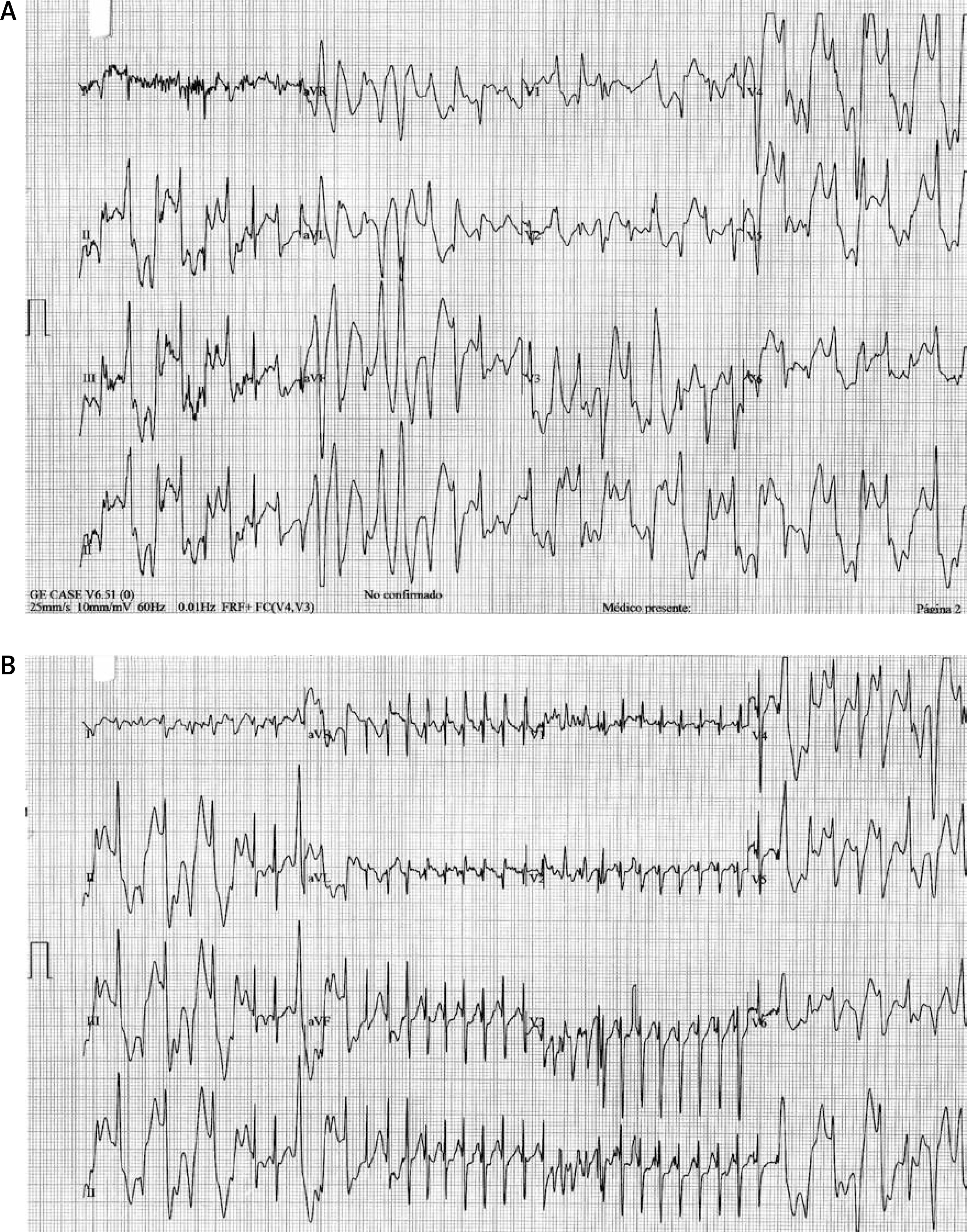
In an exercise stress test (Bruce protocol; Figure 2) a monomorphic VPB occurred when the heart rate reached 100 bpm. Ventricular premature beats in bigeminy and pleomorphic couplets were observed shortly after, then bidirectional VT at 200 bpm occurred, and a brief episode of polymorphic VT (7 beats) was recorded just before the exercise was stopped. These findings were associated with pallor, diaphoresis, and dyspnea. Paroxysmal supraventricular tachycardia was documented in the recovery phase. An electrophysiology study was performed. No ventricular or supra-ventricular arrhythmias were induced despite programmed stimulation from the right atrium, coronary sinus, and right ventricular apex (with single and double ventricular extra-stimuli at two different driven cycle lengths: standard Josephson protocol). Single nodal physiology was demonstrated. Absence of an accessory pathway was confirmed. No pharmacological challenge (isoproterenol, dobutamine) was performed.
Figure 2
Exercise stress testing. The first strip shows the polymorphic ventricular tachycardia preceded by bidirectional ventricular tachycardia (at 52 s of the first stage). The recovery phase is presented on the second strip where a non-sustained narrow QRS tachycardia is observed
With the primary diagnosis of CPVT, the patient was referred for genetic testing. Genomic DNA was extracted from whole blood. In the first instance, previous assent from the patient and permission of their parents was obtained, and an informed consent form was signed and collected after an explanation of the risks, limitations and achievements of the genetic testing, including the consideration in the future for both parents to carry out confirmatory studies, if they would have the will to participate.
Genetic testing was performed using a targeted sequence Haloplex custom design (Agilent Technologies, Santa Clara, CA, USA) including 83 cardiogenes, in a MiSeq device (Illumina, San Diego, CA, USA). Bioinformatic analyses (alignment and variant calling) were performed using BaseSpace (Illumina), and variants were annotated using ANNOVAR (http://wannovar.wglab.org/). Variants were filtered for quality, impact, and functional consequences according to SIFT (http://sift.jcvi.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Taster (http://www.mutationtaster.org/), FATHMM (http://fathmm.biocompute.org.uk/) and Provean (http://provean.jcvi.org/ index. php). The analysis identified a variant that affects function (c.11836G>A, rs794728777, or p.Gly3946Ser mutation) in the RYR2 gene in a heterozygous state. The mutation was verified in the patient and tested in both parents by Sanger sequencing in an ABI PRISM 3100 genetic analyzer (ABI 3100, Applied Biosystems, Foster City, CA). Only the patient carried the p.Gly3946Ser mutation in a heterozygous form, suggesting a de novo origin (Figure 3).
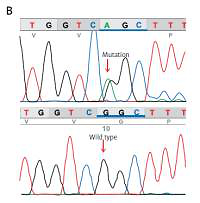
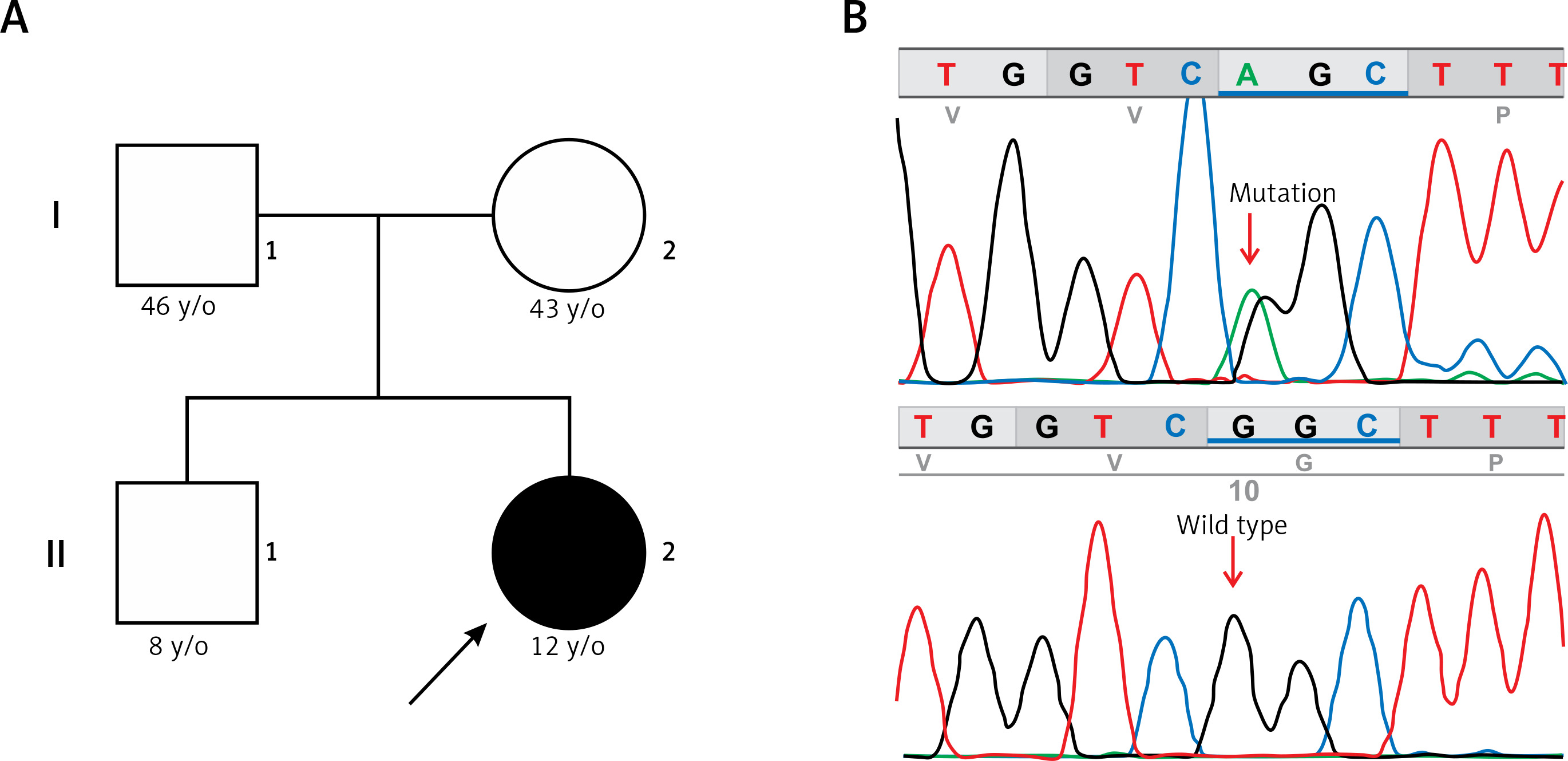
Figure 3
A – Family pedigree. Both parents and sibling are unaffected and do not carry the c.11836G<A mutation. B – Electropherogram of the proband, showing the missense c.11836G<A mutation (p.Gly3946Ser) in exon 88 of the RYR2 gene
The patient was treated with propranolol up to 1 mg/kg/day. After only 6 days of pharmacological treatment, a significant reduction in arrhythmic load was observed, with only a few isolated monomorphic VPB and couplets in Holter monitoring (276 VPB/24 h (0.3%)). Longer exercise time was achieved on a second exercise stress test (with the same Bruce protocol); VPB and bigeminy were only observed after the heart rate reached 120 bpm. The test was terminated when the first couplet was identified, at the first minute of phase 4 (for energy consumption of 12 MET). The patient experienced no symptoms during exercise. At 57-month follow-up, the girl remains asymptomatic and with no recurrence of syncope or palpitations.
To date, mutations in four different genes have been reported to cause CPVT [5], and also in the RYR2 gene (encoding the cardiac ryanodine receptor channel) [6, 7] mutations are found in 60–65% of CVPT patients and are transmitted in the autosomal dominant pattern. CALM1 (encoding calmodulin) [8], TRDN (encoding triadin) [9], or CASQ2 (encoding cardiac calsequestrin) [10] may also be the cause of CPVT.
The key feature underlying pathogenesis of CPVT is the aberrant release of Ca2+ into the sarcoplasmic reticulum during diastole (termed a transient inward current) leading to diastolic Ca2+ leak, which provides a substrate for delayed afterdepolarizations (DADs), specifically in the setting of β-adrenergic stimulation during stress or exercise. It has been proposed that CPVT-causing RYR2 mutations result in a gain-of-function of the ryanodine receptor leading to a diastolic Ca2+ leak [2, 11, 12].
Massive parallel sequencing helped identify a de novo missense RYR2 mutation c.11836G>A (p.Gly3946Ser) in the patient (Figure 3 B), located in one of the mutational hotspots of this gene. Annotation showed that the variant is predicted as damaging or deleterious by more than 5 in silico functional prediction programs. It is considered as pathogenic in the ClinVar database (www.ncbi.nlm.nih.gov/clinvar/variation/201315/), and has been previously reported in other patients with CPVT [7]. Although the mutation has not been functionally characterized in vitro, it is considered as pathogenic according to the criteria of Campuzano [13].
The case here reported exemplifies the clinical picture of CPVT in a child who suffered exercise-induced syncope misdiagnosed as seizures. She had a history of multiple falls while cycling. Initially, her parents were not concerned, believing the child was unskilled, but became concerned when they observed tonic extremity movements during the falls. As frequently occurs in hereditary sudden cardiac death syndromes, the child was misdiagnosed with epilepsy and treated with anticonvulsant drugs, showing no clinical response [14]. Between 20% and 30% of patients undergoing long-term follow-up in hospital epilepsy clinics do not have epilepsy [15]. In this case a more detailed initial clinical evaluation would have revealed that pseudoseizures were indeed related to exercise and raise suspicion of a cardiac syncope. It has been reported that a noninvasive cardiovascular evaluation including head-up tilt test, carotid sinus massage electroencephalography, and blood pressure monitoring could identify an alternative diagnosis in up to 41% of patients with apparent epilepsy, mainly vasovagal syncope [16]. However, confirming that convulsive syncope is the result of a cardiac arrhythmia is not an easy task. Previous observational studies and case reports have shown that prolonged QT and Brugada syndrome should be suspected in children who experience seizures for the first time.
The present case indicates that this is also true for CPVT [17]. The neurological investigation is insufficient if it does not include cardiological evaluation [18, 19] as previously stated. Failure to perform a cardiological evaluation may lead to severe consequences. Sudden death occurs in up to 33% of CPVT patients, and almost 80% experienced cardiac events before age 40 years, despite treatment [1]. Sports competition and extreme physical exercise are contraindicated in these patients. Beta-blockers are the first-line treatment and flecainide can be added if necessary. In selected cases, mainly those unresponsive to drugs, implanted cardiac defibrillator or left sympathectomy can be considered [20]. Propranolol treatment at 1 mg/kg/day led to an essential reduction of VPB (from 14% to 0.3%) in our patient. With pharmacological treatment only, she has remained asymptomatic after a 57-month follow-up.
Finally, concerning the genetic testing, the direct contact (instead of family contact) with both parents was the best method to ensure the later participation in the cascade screening after the identification of the mutation in the patient [21]. Because the mutation was found only in the index case, it most likely occurred de novo. However, gonadal mosaicism in one of the parents, while highly unlikely, cannot be wholly ruled out. Furthermore, 30% of patients with CPVT had sudden cardiac arrest as first presentation [22]. Because of this, genetic testing is useful to identify those patients with an apparent negative phenotype in cascade screening, mainly for carriers of mutations in RyR2 and CASQ2 [20]. A negative test does not exclude the clinical management in a suspected case of CPVT, even in cases of phenocopy or an unknown gene. Incidental findings can occur with the use of massive parallel sequencing, and in all cases, the results of the test must be provided in a clinical context, by healthcare professionals with expertise in the area.
In conclusion the case herein reported exemplifies (1) the diagnostic challenge of hereditary sudden death syndromes and utility of genetic testing; (2) light exercise (recreational cycling) as a trigger for a malignant arrhythmia; (3) an excellent response to β-blocker treatment alone in CPVT.