Definition of microRNAs, biogenesis and rationale for their use
MicroRNAs are small non-coding post-translational biomolecules which, when expressed, modify their target genes [1–3]. It is estimated that microRNAs regulate production of approximately 60% of all human proteins and enzymes that are responsible for major physiological processes [1, 2]. MicroRNAs are produced by a variety of cells [1–4]. In cardiovascular disease pathophysiology, there are several cell lineages that produce microRNAs, including endothelial cells (ECs), vascular smooth muscle cells (VSMCs), macrophages, platelets, fibroblasts, and cardiomyocytes [3–5]. There is a constant crosstalk between microRNAs derived from various cell sources.
MicroRNAs regulate gene expression at the post-transcriptional level by binding to 3′- or less often to 5′-untranslated regions of target messenger RNAs (mRNAs), which in consequence leads to inhibited translation and/or induces degradation of targeted mRNA [1]. Through this mechanism, a single microRNA can alter the function of multiple mRNAs. Furthermore, circulating microRNAs in the serum are resistant to lysis and are stable against RNase, as they are hidden in microparticles, apoptotic bodies, etc. [1].
MicroRNA biogenesis is quite complex [2]. The primary microRNA (Pri-miRNA) is produced in the cell nucleus through the transcription of a DNA strand mediated by RNA polymerase II [2]. After transcription, Pri-miRNA is cleaved by the enzymatic complex DROSHA into a micro-RNA precursor (pre-miRNA). Pre-miRNA is exported to the cytoplasm by exportin-5 and cleaved by Dicer (an RNA degrading enzyme) and produces approximately 22 nucleotide RNA duplexes. A microRNA strand is transferred to the Argonaute complex (AGO), forming an RNA-induced silencing complex (RISC), and guides it to pair with the target mRNA through binding of the microRNA seed sequence with the microRNA recognition site on the mRNA. MicroRNAs are secreted out of cells via exosomes [2]. Presently, more than 2000 human microRNAs have been identified (www.mirbase.org), and some of them are involved in atherosclerotic processes [4, 5].
Atherosclerosis initiation and progression are driven by multiple pro-inflammatory and pro-thrombotic microRNAs that overcome microRNAs with protective functions against atherosclerosis [4–6]. Atherosclerotic plaque rupture is the leading cause of cardiovascular death resulting from acute coronary syndromes (ACS), both in ST-segment (STEMI) and non-ST segment elevation (NSTEMI) myocardial infarction, as well as cardiac remodeling and fibrosis following ACS [4–6].
In this review, we focus on the role of cardiomyocyte-derived and fibroblast-derived microRNAs that are involved in the regulation of genes associated with cardiomyocyte function and atherosclerosis-related cardiac ischemia. Furthermore, we discuss microRNAs which can be potentially used in regenerative cardiology. To understand the regulatory functions of cardiomyocyte-derived microRNAs, it is essential to discuss the structure of cardiac cells which comprise the heart.
The cardiac muscle
The cardiac muscle is a very complex organ; however, for the purpose of this review we will simplify our discussion regarding the structure and function of cardiomyocytes and fibroblasts in the human heart. The human heart is composed of region-specific cardiomyocytes (atrial or ventricular), ECs, VSMCs, fibroblasts which produce extracellular matrix, and blood vessels [7, 8]. The cardiomyocyte (cardiac muscle cell) is the fundamental unit responsible for heart contractility [7, 8]. Cardiomyocytes are involved in the contractile function of the heart with constant contraction and relaxation during the cardiac cycle throughout an individual’s entire life span [7, 8]. Each human cardiomyocyte contains both contractile proteins, usually 1 or 2 nuclei, and large numbers of mitochondria which provide adequate levels of ATP required by the cells (Figure 1). The basement membrane of cardiomyocytes is composed of glycoproteins (laminin and fibronectin), type IV collagen, as well as proteoglycans [9]. This membrane is responsible for trapping calcium ions responsible for contractility and those that permit the mechanical and electrical coupling of adjacent cells.
Figure 1
A basic schematic diagram of human left ventricular muscle structure and cardiac muscle-derived microRNAs. A – A sarcomere is composed of myofibrils, each containing myofilaments: the thick filaments are composed of myosin, while the thin filaments are composed of actin, tropomyosin, troponin C, and troponin I. Myosin contains two heads having ATPase activity. Calcium ions (Ca2+) bind via troponin C. miR-208b and miR-499 regulate genes for myosin heavy chains. B – Cardiac muscle cells are striated, branched, involuntary, and usually contain a single nucleus. C – Fibroblasts provide support to cardiac muscle cells, as they are responsible for deposition of extracellular matrix in the heart
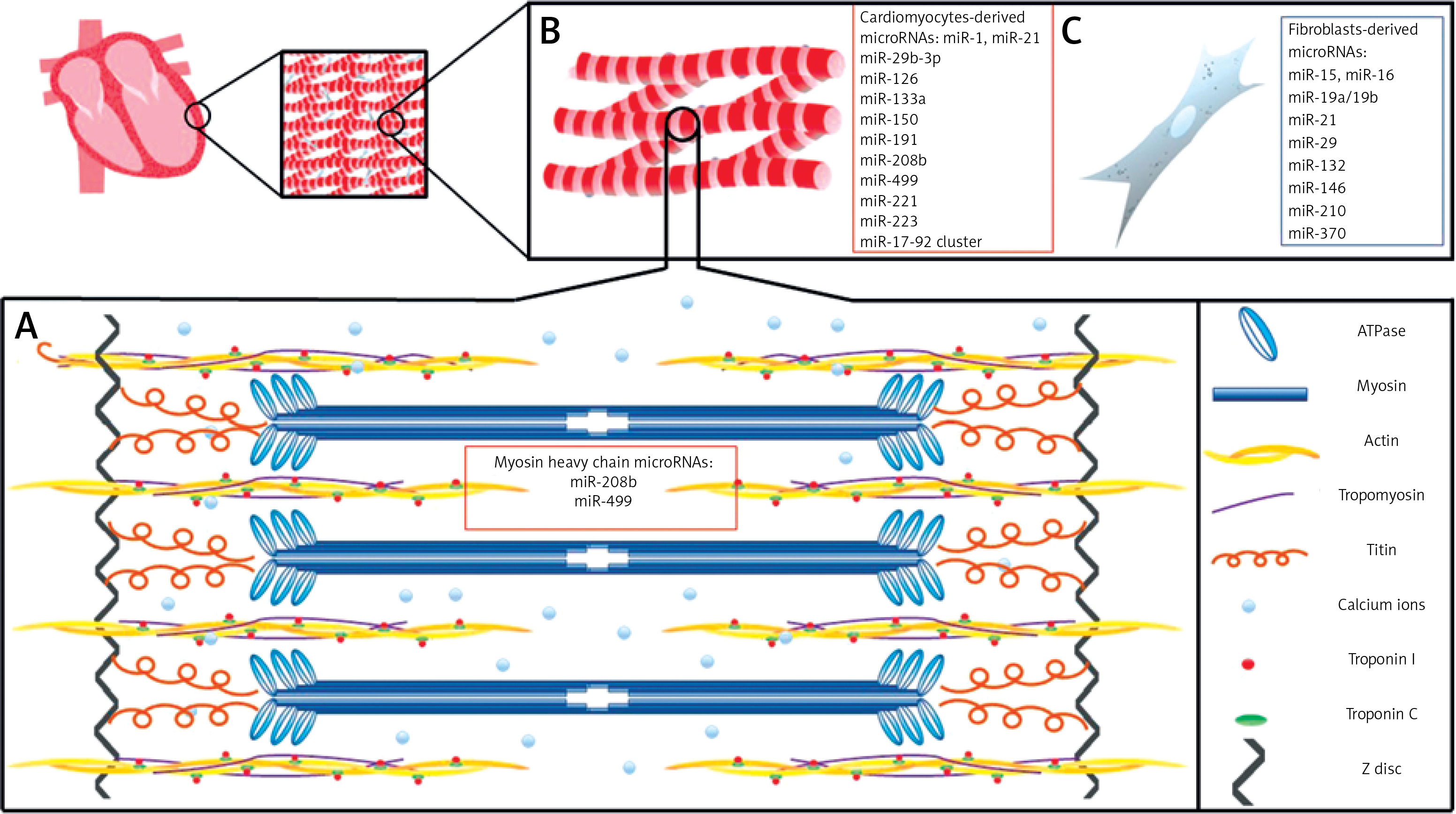
Sarcomeres, the essential elements of cardiomyocytes, are composed of contractile proteins such as actin and myosin, and cytoskeletal proteins which maintain mechanical integrity of the cell [7, 8, 10]. Sarcomeres also contain the regulatory proteins troponin and tropomyosin. Cardiac troponins (cTn) I and C control the binding of myosin to actin and consequently take part in the contraction and relaxation of cardiac muscles (Figure 1). Myosin and actin generate the force of contraction, while thin filament proteins calibrate the force generated by contractile proteins. Also, titin, a myofilament protein, functions as a scaffold for sarcomeric assembly and as a molecular spring in striated muscle cells to regulate both systolic and diastolic function [11]. Titin molecules anchor to the Z-disc and M-line of the sarcomere. Alterations in titin isoform expression and titin proteolysis contribute to contractile dysfunction in dilated cardiomyopathy, ischemic heart injury, and COVID mRNA vaccination [12].
There are differences in the composition of myosin heavy chains and in fiber arrangement between atrial and ventricular regions. The β isoform of myosin heavy chain (MHC-β) is predominant in the adult ventricles, while the α isoform (MHC-α) is predominant in the atria [8]. In addition, there is a difference in myosin light chain isoforms between the atria and ventricles [10]. All cardiomyocytes comprise the fibers of the cardiac muscle, which communicate through gap junctions. The adjacent cells are anchored together by desmosomes.
In the heart, cardiomyocytes co-exist with fibroblasts. Following cardiac injury, such as in myocardial infarction (MI), fibroblasts become activated and can transform into myofibroblasts, cells which exhibit the behavior of both a fibroblast (generating extracellular matrix) and a smooth muscle cell (ability to contract). Alternatively, they may proliferate as fibroblasts, contributing to scarring within the post-MI area.
The natural ability of cardiac muscle cells to regenerate is minimal. Studies have shown that approximately 1% of cardiac cells are renewed in young adults annually. However, this decreases to about 0.3–0.45% for individuals above 75 years of age [13]. Following cardiac injury there is an irreversible loss of cardiomyocytes and intense replacement of the cardiomyocyte space by excessive fibroblastic proliferation, which leads to scarring and post-MI acute and chronic complications.
Despite the progress which has been made in regenerative therapy in cardiology, such as stimulation of human pluripotent stem cell-derived cardiomyocytes or human embryonic stem cells, there is still room for further development of novel technologies in the prevention of adverse cardiac outcomes [10]. One possible direction is the employment of microRNAs that naturally regulate all physiological and pathophysiological processes in the heart [14].
MicroRNAs and heart development
MicroRNAs regulate all stages of embryonic cardiac development and heart functions throughout an individual’s life [15]. This process is highly conserved in mammals [16]. The heart is the first organ which starts to develop, usually in the third week of gestation [16]. MicroRNAs appear during heart development at different stages and have diverse functions (Table I). During heart development, cardiomyocyte structure and proliferation are regulated by 2 critical microRNAs: miR-1 and miR-133 [17]. Their activity must be well balanced, as either over-expression or under-expression is associated with defects [18]. Studies suggest that miR-1 may regulate sarcomere formation in the mammalian heart [19]. A study involving a miR-133a double-knockout embryonic heart showed increased cardiomyocyte proliferation as well as apoptosis, disrupted sarcomere structure, and dysregulated expression of smooth muscle genes [20]. In the early stages of heart development, miR-128a stimulates the differentiation of cardiomyocyte progenitor cells into various subtypes of cardiomyocytes by modulating the differentiation of cardiac progenitor cell populations [21]. A cluster of miR-17-92 promotes cardiomyocyte proliferation in embryonic, postnatal, and adult hearts, whereas members of the miR-15 family decrease cardiomyocyte proliferation and induce apoptosis [22–24]. Several microRNAs regulate the expression of cardiac myosin genes, including miR-208b (the most important) and miR-499 (dependent), which are involved in the regulation of genes responsible for producing myosin heavy chains [25]. Additionally, one study found that miR-499 was an important regulator of ventricular specification [26]. Expression of these microRNAs is also crucial in postnatal life; however, high expression levels of miR-208b do not lead to cardiomyocyte proliferation, but rather to cardiomyocyte hypertrophy, atrial fibrillation, and heart failure [27].
Table I
MicroRNAs involved in heart development (data from mouse studies, or human induced pluripotent stem cells) and their function
| MicroRNAs involved in heart development | Subject of study | Activity/significance | Reference |
|---|---|---|---|
| miR-1 | Mice | Promotes differentiation of embryonic stem cells into cardiomyocytes, regulates sarcomere formation in the mammalian heart, cardiomyocyte structure, proliferation, and cardiac conduction | [18, 19] |
| miR-133a | Mice | Inhibits differentiation of embryonic stem cells into cardiomyocytes, regulates sarcomere structure, cardiomyocyte proliferation and apoptosis, expression of smooth muscle genes | [20] |
| miR-128a | hiPSCs | Subtypes of cardiomyocytes, modulates differentiation of cardiac progenitor cell populations | [21] |
| A cluster of miR-17-92 | Mice | Promotes cardiomyocyte proliferation in embryonic, postnatal, and adult hearts | [22, 23] |
| miR-15 miR-16 | Mice | Decreases cardiomyocyte proliferation and induces apoptosis | [24] |
| miR-499 | Mice | Production of myosin heavy chains, ventricular specification | [25, 26] |
| miR-208b | Mice | Production of myosin heavy chains | [27] |
| miR-145 | Mice | Differentiation of multipotent cardiac neural crest stem cells into VSMCs | [15] |
| miR-21 miR-31 miR-103/107 miR-155 miR-200 | Mice | Implicated in epicardial development | [15] |
| miR-218 | Zebrafish | May control cardiac cell migration | [28] |
| miR-143/145 | Mice | May be responsible for cell-cell contact | [29, 30] |
It is possible that miR-218 may control cardiac cell migration, whereas miR-143 (well-known for VSMC switching) may be responsible for cell-cell contact; however, this has not yet been confirmed in mammals [28]. In the early stages of murine cardiogenesis, miR-143 and miR-145 are highly expressed and are transcribed as a bicistronic cluster [29, 30]. MicroRNAs, such as miR-145, are involved in the differentiation of multipotent cardiac neural crest stem cells into VSMCs during heart organogenesis. Moreover, miR-145 is very important as it is capable of reprogramming adult fibroblasts into VSMCs [31]. MicroRNAs, including miR-21, miR-31, miR-103/107, miR-155, and the miR-200 family, have been implicated in epicardial development [15].
All embryonic microRNAs act through their target genes, resulting in synthesis of proteins [32–70]. The microRNAs which appear to play a pivotal role in cardiac development include miR-1 (by promoting myogenesis and MHC differentiation) and miR-133 (by promoting mesoderm formation at the early stages of embryological development) (Table II) [32–35]. The expression of miR-1 and miR-133 in the embryonic heart is regulated by transcription factors SRF, MEF2, myogenic regulatory factor (MRF), and MyoD [34, 35]. In addition, miR-208b together with miR-499 drives muscle cell specification and slows MHC upregulation through targeting β-MHC and Sox6 genes [36–39].
Table II
Target genes associated with cardiomyocyte-derived microRNAs
| microRNA | Target gene | Activity/significance | Reference |
|---|---|---|---|
| miR-1 | HDAC4, MEF2C | [19, 32] | |
| miR-133a | SRF | [20, 32, 34] | |
| miR-128a | Isl1, Sfrp5, Hcn4, Irx4 | [21, 57, 58, 59] | |
| cluster of miR-17-92 | PTEN, CDK1 | Regulation of cardiomyocyte proliferation in embryonic, postnatal, and adult hearts | [23, 60, 61] |
| miR-15 miR-16 | C/EBPβ, IGF1 | Protection/downregulation of cardiac hypertrophy | [54] |
| miR-208b | β-MHC, Sox6, Purβ, Sp3, HP1β | [25] [37] | |
| miR-499 | Sox6, cyclin D1 | Inhibition of cell proliferation and promotion of cell apoptosis in P19CL6 cells and cardiomyocytes | [38] [39] |
| miR-133b | PTBP1, TAGLN2 | Alleviates apoptosis and cardiac fibrosis, modulates collagen deposition | [53] |
| miR-143/145 | HK2 | Addresses G6PD, PPP pathways and p62. Activation of these signaling pathways induces a reductive redox shift, resulting in cardiomyopathy | [55] |
| miR-155 | NEDD4 | Cardioprotective against cardiomyocyte apoptosis; modulates myofibroblast density | [56] |
| miR-218 | REST | Regulates cardiac differentiation of embryonic stem cells by regulating the Wnt/β-catenin signaling pathway and GATA4 | [63] |
| miR-124 | STAT3 | [64, 65] | |
| miR-21 | Ajuba/Isl1 | Differentiation of BMSCs into cardiomyocyte-like cells; contributes to formation and maintenance of cell-cell connections and supports cell division and migration | [66] |
| miR-31 | CAMK2D, YWHAE | Promotes loss of dystrophin and nNOS in human | [67] |
| miR-103/107 | PANKs, FADD | Modulates mitochondrial concentration; regulates systemic glucose metabolism; H2O2-induced necrosis | [68, 69] |
| miR-200b | GATA-4 | Transcription factor; regulation of developmental processes of the heart, cardiac myocyte proliferation, differentiation and survival | [70] |
[i] ANP – atrial natriuretic peptide, BMSCs – bone marrow-derived mesenchymal stem cells, CAMK2D – calcium/calmodulin dependent protein kinase II delta, CDK1 – cyclin dependent kinase 1, C/EBPβ – CCAAT enhancer binding protein beta, ESCs – embryonic stem cells, FADD – Fas-associated via death domain, G6PD – glucose-6-phosphate dehydrogenase, GATA4 – GATA binding protein 4, Hcn4 – hyperpolarization activated cyclic nucleotide gated potassium channel 4, HDAC4 – histone deacetylase 4, HK 2 – hexokinase 2, HP1β – heterochromatin protein 1β, IGF1 – insulin-like growth factor-1, Irx4 – iroquois homeobox 4, MEF2 – myocyte-specific enhancer factor 2, MHC – myosin heavy chains, mRNA – messenger ribonucleic acid, MyoD – myoblast determination protein 1, NEDD4 – neural precursor cell expressed developmentally down-regulated protein 4, nNOS – neuronal nitric oxide synthase, PANKs – pantothenate kinases, PPP – pentose phosphate pathway, PTBP1 – polypyrimidine tract-binding protein 1, PTEN – phosphatase and tensin homolog, Purβ – purine rich element binding protein B, REST – RE1 silencing transcription factor, Sfrp5 – secreted frizzled related protein 5, Sox6 – SRY-box transcription factor 6, Sp3 – specificity protein 3, SRF – serum response factor, STAT3 – signal transducer and activator of transcription 3, TAGLN2 – transgelin 2, TnT – troponin T, YWHAE – tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein epsilon, Wnt – wingless/integrated.
During embryogenesis, the developmental potential of cells is gradually restricted as they become modified toward a specific function. As cell development continues, its function is determined by DNA methylation and chromatin remodeling, which is mostly regulated by transcription factors [40–44]. Cardiomyocytes being derived from pre-cardiac mesoderm are modulated by transcription factor MESP1 as well as other dependent factors, including GATA4, MEF2C, SRF, or ISL1 [40–44]. Different microRNAs target their specific genes, thus modifying the transcription factors and regulating cardiomyocyte development [40]. The molecular mechanisms regulating cardiomyocyte proliferation include core cell cycle proteins such as cyclins (cyclin D1, D2, B1), cyclin-dependent kinases (CDK1, CDK4), and nuclear proteins which regulate the cell cycle, such as Gata4, Meis1, and Hand2 [40–44]. In addition, signaling pathways including Hippo, Notch, and p38 kinase pathways are involved in cardiomyocyte proliferation [45–51]. Cardiomyocyte proliferation may also be stimulated by exogenous factors such as hypoxia or inhibited by thyroid hormones [52, 53].
MicroRNAs act through their target genes, and in this way are involved in the process of heart regeneration. After myocardial damage, the loss of cardiomyocytes is mostly irreversible, which results in scar formation and cardiac remodeling [53, 71, 72]. In this process, microRNAs can influence cardiac hypertrophy [54], especially by modulating glucose-6-phosphate dehydrogenase or pentose phosphate pathways, by stimulating cardiac cell apoptosis and subsequent fibrosis [53], or by modifying myofibroblast density [55, 56]. Tracing different cell lines and their potential to survive and regenerate after myocardial injury is the main issue of many current studies, as it can result in establishing genes or their target proteins as potential therapeutic points.
The fetal heart is an excellent model for the synchronized, often overlapping microRNA activity during development of the mature heart. To understand fibroblastic substitution following cardiomyocyte death in cardiac ischemia, it is important to elucidate the mechanisms involved in embryonic heart maturation. In regenerative cardiology, using microRNAs to stimulate fibril production in cardiomyocytes, as well as keeping cardiomyocytes in situ (homing), is challenging. In addition, microRNA-based inhibition of excessive autophagy and apoptosis of cardiomyocytes should be well balanced. Finally, stimulation of fibroblasts into induced cardiomyocytes (iCMs) requires regulation of microRNAs.
Acute cardiac ischemia
Diagnostic microRNAs
Cardiomyocyte-derived microRNAs diagnostic for ACS
MicroRNAs contained in cardiomyocytes are released into the blood stream as a result of cardiac cell ischemia, leading to cell damage [73–76]. Among the cardiomyocyte-enriched microRNAs, miR-1, miR-21, miR-133a, miR-133b, miR-145, miR-208b, miR-223, and miR-499 are considered markers of myocardial damage and have been studied in the context of possible diagnostic biomarkers for ACS [77–80]. These microRNAs are detectable in the serum, often before traditional markers of cardiac necrosis are detectable (Table III). Thus, microRNAs may be used to differentiate patients with stable angina from vulnerable patients. Some microRNAs might be better than others as they are characterized by their high diagnostic accuracy and rapid release preceding cardiac troponins (cTn) [76]. Studies showed that the diagnostic sensitivity and specificity of miR-499 was very high (between 98 and 100%) [77, 78]. In a study by Abdou et al., diagnostic accuracy (AUC) for miR-499 was 0.90 (95% CI: 0.845–0.955) with 89% sensitivity and 83% specificity in STEMI patients [79]. Furthermore, 74% of patients had a significant increase in miR-499 expression levels compared to 65% positive cTn in the first 3 h of chest pain [79]. A meta-analysis of 14 observational studies showed that circulating miR-499 is a reliable biomarker for diagnosing acute myocardial infarction in patients [80]. Additionally, upregulation of miR-208a can be easily detected in 100% of ACS patients within 4 h of the onset of symptoms, whereas it remained undetectable in non-ACS patients [81]. A meta-analysis of 13 studies by Wang et al. involved a group of 1703 patients with ACS and a group of 1589 control patients [82]. They found that miR-208b is characterized by a high diagnostic accuracy for ACS (AUC = 0.93 (95% CI: 0.91–0.95), p < 0.001), with a pooled sensitivity of 83% and specificity of 97% [80]. All the microRNAs are placed in close proximity to the myosin and actin apparatus; thus, their high expression levels are associated with a greater loss of myofibrils, leading to an unfavorable reduction in left ventricular ejection fraction and post-MI remodeling [82–84]. An interesting issue is whether microRNAs can be considered potential biomarkers of acute coronary artery occlusion in an infarct-related artery (IRA). Occlusion of an IRA occurs both in STEMI and NSTEMI-ACSs [85]. A recent study by Gacoń et al. showed promising results for miR-133b and miR-124, as they were associated with an increased risk of acute coronary artery thrombosis [85].
Table III
Cardiomyocyte-derived microRNAs diagnostic for acute coronary syndrome and prognostic for adverse cardiovascular events following acute coronary syndrome
| microRNA | Period | Subject to study | Activity/significance | Ref. |
|---|---|---|---|---|
| Diagnostic for ACS | Study group(s) | MicroRNAs with the highest clinical diagnostic potential AUC (95% CI), p-value | ||
| miR-208b miR-499 | 80 ACS patients and 50 healthy controls | Human serum | High diagnostic potential for ACS: miR-208b: 0.9996 (0.9983–1.001), p < 0.001 with a sensitivity of 98% and specificity of 100% miR-499: 0.9992 (0.9970–1.001), p < 0.001 with a sensitivity of 98% and specificity of 100% superior to cTn: 0.940 (0.8742–1.006), p < 0.001 | [77] |
| miR-1 miR-133 miR-208 miR-499 | 145 NSTEMI patients and 30 healthy controls | Human serum | High diagnostic potential for NSTEMI-ACS: miR-1: 0.7731 (0.6865–0.8597), p < 0.05 miR-133: 0.928 (0.8857–0.9704), p < 0.05 miR-208: 0.994 (0.9869–1.001), p < 0.05 miR-499: 0.9945 (0.9863–1.003), p < 0.05 compared to cTn: 0.778, p < 0.05 | [78] |
| miR-499 | A meta-analysis of 14 studies including 1989 patients with ACS and 1732 control patients | Human serum | High diagnostic potential for ACS: miR-499: 0.98 (0.96–0.99), p < 0.001 with a pooled sensitivity of 84% and specificity of 97% | [80] |
| miR-208b | Meta-analysis of 13 studies including 1703 patients with ACS and 1589 control patients | Human serum | High diagnostic potential for ACS: miR-208b: 0.93 (0.91–0.95), p < 0.001 with a pooled sensitivity of 83% and specificity of 97% | [82] |
| miR-133b miR-124 | 27 patients with an occluded infarct-related coronary artery ACS vs. 16 patients with patent artery | Human serum | Good diagnostic potential for infarct-related coronary artery occlusion: miR-133b: 0.704 (0.547–0.860), p = 0.006 with a sensitivity of 63% and specificity of 81.3% miR-124: 0.787 (0.649–0.924), p < 0.001 with a sensitivity of 59.3% and specificity of 87.5% | [85] |
| Prognostic for MACCE after ACS | ||||
| miR-197 miR-223 | 340 patients with ACS | Human serum | Prognostic for CVD after ACS at 4 years miR-197: 2.24 (1.25–4.01), p = 0.006 miR-223: 4.94 (1.42–17.2), p = 0.012 | [86] |
| miR-208b | 21 patients with ACS | Human serum | Prognostic for CVD at 6 months miR-208b: 5.08 (1.13–22.82) p = 0.03 | [87] |
| miR-223-3p | 598 ACS patients randomized to ticagrelor vs. prasugrel treatment | Human plasma | miR-223 levels were prognostic for CVD/re-MI/IS at 30 days: 15.74 (2.07–119.9), p = 0.008 and at 1 year: 3.18 (1.40–7.19), p = 0.006 | [88] |
| miR-1-3p | 142 patients with ACS or stroke | Human serum | Prognostic for CVD at 6 years miR-1-3p: 2.73 (1.22–6.12), p = 0.014 | [89] |
| miR-34a miR-208b | 359 patients with ACS | Human serum | Prognostic for CVD and left ventricular remodeling (LVEDV increase > 10%) at 6 months. miR-34a: 17.91 (2.07–98.81), p = 0.003 miR-208b: 4.18 (1.36–12.83), p = 0.012 | [90] |
| miR-150 | 113 patients with ACS | Human serum | Prognostic for left ventricular remodeling. (LVEDV increase > 10%) at 1 year miR-150: 1.233 (1.125–1.352) | [91] |
| Responsive to antiplatelet therapy | ||||
| miR-223-3p | 62 patients with NSTEMI | Human serum | Decreased circulating miR-223 is a marker of lower response to clopidogrel in NSTEMI. 0.111 (0.018–0.692), p = 0.019 | [99] |
| miR-223-3p | 21 male patients with NSTEMI taking clopidogrel (n = 11) or prasugrel/ticagrelor (n = 10) in addition to aspirin | Plasma serum | Plasma miR-223 was elevated with decreasing platelet reactivity | [101] |
| miR-21 miR-221 miR-223 | 272 ACS patients, including 21 extremely high responders and 18 extremely low responders | Human serum | miR-223, miR-221, and miR-21 are independently associated with clopidogrel antiplatelet responsiveness in ACS patients. However, the association could be influenced by the interaction with CYP2C19*2 genotype | [102] |
| Preventive of in-stent restenosis | ||||
| miR-22 | Self-healable spongy coating stent with miR-22 | Minipigs | PCEC@miR-22 coated stents showed reduced inflammation, low switching of VSMC phenotype, and low secretion of extracellular matrix, which significantly inhibited in-stent restenosis | [107] |
| miR-21 | Animal model: suppression of vascular miR-21 correlated dose dependently with reduced luminal obliteration. Furthermore, anti-21 did not impede re-endothelialization | Rats | Compared with bare-metal stents, anti-21-coated stents effectively reduced in-stent restenosis. However, systemic anti-miR-21 had substantial off-target effects, lowering miR-21 expression in liver, heart, lung, and kidney with a concomitant increase in serum creatinine levels | [108] |
| miR-30b-5p | Detection of restenosis risk | Human serum | Downregulation of miR-30b-5p was predictive for coronary in-stent restenosis | [109] |
[i] ACEi/ARB – angiotensin-converting enzyme inhibitors/angiotensin receptor blockers, ACS – acute coronary syndrome, AUC – area under the curve, CI – confidence interval, CVD – cardiovascular death, cTn – cardiac troponins, hiPSCs – human induced pluripotent stem cells, IS – ischemic stroke, LVEDV – left ventricular end diastolic volume, MACCE – major adverse cardiac and cerebral events, MI – myocardial infarction, MRA – mineralocorticoid receptor antagonists, VSMCs – vascular smooth muscle cells.
Prognostic microRNAs
MicroRNAs are prognostic for adverse cardiovascular events following ACS.
MicroRNAs associated with acute cardiac ischemia are often biomarkers of major cardiac and cerebral events following ACS [86–94]. Decreased miR-150 and increased serum levels of miR-1, miR-34a, miR-223, miR-146a, miR-197, and miR-208b are associated with increased cardiovascular mortality following ACS, major adverse cardiovascular events, heart failure (HF), and left ventricular remodeling [86–92] (Table III). In a study by Schulte et al. which included 340 ACS patients, levels of miR-197 and miR-223 were predictive for cardiovascular death (CVD) [86]. In a study by Hromadka et al., adding miR-223-3p into the model for calculating ischemic risk following ACS significantly increased the predictive accuracy for CVD (OR = 10.8, 95% CI: 1.37–85.07, p = 0.024), as well as the combined ischemic endpoint (CVD/re-MI/IS) within 30 days and 1 year [88]. In a study by Lin et al., only the level of miR-150 differed between patients who developed HF (HR = 1.233; 95% CI: 1.125–1.352) from those who did not at a 1-year follow-up [90]. Some researchers have suggested that inflammation-related microRNAs such as miR-145, miR-146a, and miR-342 can serve as biomarkers of adverse prognosis following ACS [91, 92].
microRNAs modifying platelets’ activity
Platelet response to dual antiplatelet treatment
A variety of therapeutic strategies were employed to diminish the risk of adverse cardiac events following ACS [95, 96]. The most important strategy includes urgent coronary percutaneous intervention (PCI) with restoration of IRA patency, preferably with provisional stenting [95, 96]. Undergoing this procedure in a timely manner limits the post-MI infarct size area, scarring, and incidence of malignant arrhythmia and episodes of HF.
Since PCI with stent implantation requires the usage of dual antiplatelet treatment (DAPT) for a prolonged period of time, it is associated with an increased risk of bleeding, particularly in elderly patients who also use anticoagulants for atrial arrhythmia [97]. MicroRNAs associated with platelet aggregation could be used as biomarkers of platelet response to DAPT treatment (Table III) [98–100]. Due to the limited value of platelet reactivity tests, this may offer a practical approach [75, 98]. MicroRNA-driven dosages of antiplatelet agents could enable personalization of antiplatelet treatment to prevent episodes of excessive bleeding or stent thrombosis [98–100]. High platelet reactivity during treatment is associated with decreased expression levels of circulating miR-233 [99]. In contrast, increased expression levels of miR-223 are associated with increased DAPT responsiveness [101]. In line with these observations, the expression of miR-21, miR-221, and miR-223 in platelets from ACS patients appeared to be decreased in non-responders who were receiving aspirin and clopidogrel [102]. Because results are not entirely consistent throughout the studies, investigations on the expression levels of microRNAs (such as miR-223 and miR-126) and properties of platelet coagulability are still ongoing [102].
MicroRNAs in the prevention of in-stent restenosis
The main drawback of stent implantation is stent restenosis [103, 104]. Data indicate that this complication could be overcome using coated stents with microRNA agonists (miR-22) or antagonists (anti-miR-21) (Table III) [105–108]. A study by Gutierrez-Carretero et al. found that low plasma levels of miR-30b-5p may have diagnostic potential for determining the risk of in-stent restenosis in coronary arteries [109].
Therapeutic approach based on microRNAs and their ability to modify target genes
Challenges in microRNA delivery
MicroRNAs target sites in mRNA mostly lie within the 3′-UTR and less frequently in 5′-UTRs or coding regions [1]. Natural microRNAs are stable and resistant to enzymatic lysis because they are hidden inside microvesicles and microparticles; therefore they are protected from quick enzymatic lysis [110, 111]. In contrast, exogenous microRNAs must obtain properties that would make them resistant to rapid lysis and increase their cellular adhesion, so they could exert their therapeutic actions. Clinical studies on micro-RNA-based therapies are feasible because novel technologies of microRNA delivery and encapsulation are developing. To decrease the expression of a specific microRNA, specific antisense oligonucleotides (ASOs), small interfering RNA (siRNA), and miRNA sponges are utilized [110–113]. ASOs (chemically modified) are preferred over natural oligonucleotides to reduce nuclease sensitivity and rapid renal clearance, but also to optimize delivery [110–112]. Genetic knockout and synthetic microRNA mimics or pre-miRNA in viral vehicles are used to enhance a specific microRNA level. Encapsulation includes adenovirus, lentivirus, AAV, microspheres, lipid nanoparticles, and liposomes [110–113].
ASOs for anti-miRs (antagomirs) are delivered as a single strand and do not seem to become part of the RISC, whereas mimics are usually applied as double strands to make them a substrate for Dicer and promote integration of one strand into the RISC. Agomir and antagomir are more stable in the blood stream, having a lasting effect between 1 and 6 weeks.
Also, various routes of microRNAs administration are used: subcutaneous injection, intravascular delivery, direct injection to the coronary circulation or into the heart, and devices coated with microRNAs. E.g. microRNA mimics of 4 cardiomyocyte-derived microRNAs are injected directly into the heart using nanocomplexes of branched polyethyleneimine-coated nitrogen-enriched carbon dots [114].
In the near future, further progress regarding the biomolecule mediated delivery of microRNAs is likely to be achieved by the development of aptamer conjugates [113]. Aptamers are single-stranded nucleic acids with high-affinity ligands of cellular surface receptors, facilitating their intracellular uptake by receptor-mediated transport [113]. They may be easily produced by standard in vitro synthesis techniques and able to be coupled to corresponding microRNA therapeutics by simple sticky-end annealing. Aptamer-conjugated microRNA agents, such as the GL21.T-miR-34c conjugate, are currently being tested in preclinical studies for selective targeting of tumor cells, including lung cancer cells.
MicroRNA-based strategies to prevent cardiac remodeling after ACS
Inhibition of extensive fibroblast proliferation resulting in scar formation
Extensive fibroblast proliferation after ACS leads to excessive left ventricle wall scarring that aggravates post-MI cardiac remodeling. microRNAs involved in the pathogenesis of post-MI left ventricular remodeling and HF are mostly fibroblast-derived [115]. Cardiac fibrosis is an interplay between pro-fibrotic microRNAs (miR-9, -15, -21, -26a, -34a, -92, -125b, 132, -155, 199b, -208a, -223, -433, and let-7c) and anti-fibrotic microRNAs (miR-7a/b, -19a, -19b, -22, -24, -29a, -29b, -30a-5p, -99, -101, -133a, -144, 146, -150, -210, -214, -370, and miR-384) [115]. Currently, microRNA-based strategies to prevent adverse outcomes following ACS are being investigated, focusing mainly on the inhibition of pro-fibrotic microRNAs. microRNA antagomirs, at least in theory, should lead to the salvage of cardiac function through decreased fibroblast proliferation
Ongoing and terminated studies involving microRNA-based treatment in ACS patients
Before a micro-RNA-based treatment can be commenced, a microRNA has to come a long way, from bench genetic studies, cell cultures, animal models (mouse, pig, monkey) into first-in-human studies [110–113]. In the cardiovascular setting, a good example of a microRNA that makes a fast tract career is miR-132-3p. Antagomir against miR-132-3p (known as CDR132L) prevented post-MI cardiac remodeling, which was confirmed in mouse models of HF, and later confirmed in pig models with ACS [110, 116, 117]. A phase I clinical trial in humans revealed good tolerability and evidence of a therapeutic benefit, such as preserved cardiac function and reversed cardiac remodeling, in HF patients (Table IV) [118]. The antagomir against miR-132-3p entered a phase II clinical study in June 2022 (https://clinicaltrials.gov/ct2/show/NCT05350969) [110]. The antagomir against miR-132-3p is among a few microRNAs showing promise for their clinical utility. The other promising microRNA in cardiac ischemia and post-MI remodeling, potentially therapeutic, is antagomiR-92a, which was found to reduce infarct size in mouse and pig models, and enhanced blood vessel growth and functional recovery of damaged tissues [116, 119, 120]. In a model of reperfused MI, a single dose of anti-miR-92a prevented cardiac remodeling without adverse effects in a porcine model [120].
Table IV
Review of candidate microRNAs tested as potential therapeutics in cardiac ischemia animal studies, phase I and phase II clinical trials in humans
| microRNA | Role, rationale for use from previous research data | Studies in human | Randomized- controlled trial | Stage of the studies (the highest available) | Route of administration | Study design | Positive/negative/ limitations | Ref. |
|---|---|---|---|---|---|---|---|---|
| miR-132-3p | Inhibition of miR-132 prevents cardiac fibrosis, and cardiac remodeling | Yes | Yes ASO antagomir against miR-132-3p (CDR132L) | Phase Ib (NCT04045405), 28 patients, completed HF-REVERT Phase II (NCT05350969), ongoing since June 2022 | i.v. (0.32, 1, 3, and 10 mg/kg) three i.v. infusions 28 days apart, 5 mg/kg, vs. 10 mg/kg, vs placebo | Chronic HF (LVEF ≥ 30% and < 50% or NT-proBNP > 125 ng/l at screening 280 patients with HF and reduced LVEF after MI. Designed to evaluate the safety and efficacy of CDR132L | Completed. CDR132L was safe and well tolerated, confirmed linear plasma pharmacokinetics with no signs of accumulation, and suggested cardiac functional improvements Ongoing (recruiting patients) | [118] [110] |
| miR-103/107 | Inhibition of miR-103/107 leads to improved glucose homeostasis and insulin sensitivity Inhibition of miR-103/107 reduces infarct size and prevents cardiomyocyte necrosis | Yes No | Yes miR-103/107-3p inhibitor (AZD4076 vs. placebo) No | Phase I (NCT02612662) Phase I/IIa (NCT02826525) 14 participants (early termination) ASO antagomiR-103-107 Healthy adult mice | s.c. 2 ml vs. placebo s.c. Injected at dose 40 mg/kg/day for 6 weeks | Healthy male subjects with NASH Patients with diabetes type 2 and NAFLD Chronic treatment with antagomiR-103 and -107 | Active, not recruiting (enrolled 40 participants) Immune-related side effects (halted for strategic reasons), off-target actions AntagomiR-103 and -107 decreased LVEF, cardiomyocyte size, and mitochondrial oxidative capacity in the absence of pathological stimuli. These data raise concern about the possible cardiac implications of the systemic use of antagomiR-103 and -107 in the clinical setting | [123] [124] [125] |
| miR-15-5p | Inhibition of miR-15b prevents cardiac remodeling | No | No LNA-modified anti-miR-15 | Preclinical stage: mice, pig models | iv. | Mice and pig after ischemia-reperfusion surgery | Anti-miR-15 in mice reduces infarct size and cardiac remodeling and enhances cardiac function in response to MI | [127] |
| miR-16-5p | Loss of miR-15 and miR-16 is associated with unsuppressed growth in preclinical models of malignant pleural mesothelioma Suppression of miR-16 protects rat hearts from ischemic injury | Yes Yes | Yes a double-stranded miR-16 mimic (TargomiRs) No | Phase I (NCT02369198) completed Case-control study (12 healthy controls and 12 ICM patients | iv. Plasma samples | Malignant pleural mesothelioma Non-small cell lung cancer Plasma samples, AC16 Cell culture and transfection with miR-16 inhibitor | Acceptable safety profile and early signs of activity of TargomiRs in patients with malignant pleural mesothelioma Inhibition of miR-16-5p has potential as a therapeutic approach to protect the heart against oxidative stress-induced injury | [128] [129] |
| miR-17-5p | Inhibition of miR-17 Inhibition of miR-17 prevents cardiomyocyte apoptosis, reduces infarct area | Yes No | Yes ASO anti-miR-17 (RGLS4326) No (Ad-Antagomir-17) | Phase Ib (NCT04536688) 19 participants (early termination) Rat study | s.c. Intramyocardial injection | Autosomal dominant polycystic kidney disease MI model rats | An interim analysis showed lack of efficacy MiR-17-5p silencing decreases the rate of apoptosis and repairs vascular injury | [130] [131] |
| miR-21-5p | Inhibition of miR-21 prevents Alport nephropathy progression Inhibition of miR-21 retains cardiac function Injection of miR-21-enriched EVs restores cardiac function | Yes No No | Yes, ASO anti-miR-21 (lademirsen) No LNA-antimiR-21 No | HERA study Phase II (NCT02855268) 43 patients (closed) Pig study Mouse study | s.c. once a week vs placebo Intracoronary i.v. miR-21-enriched derived from Hek293T cells | Alport syndrome | Interim analysis showed no meaningful improvement in kidney function compared with placebo In a pig model of HF, reduced cardiac fibrosis, hypertrophy and improved cardiac function Restores cardiac function in mice after MI | [132] [133] [134] [135] |
| miR-22-3p | Inhibition of miR-22-3p improves cardiac function | No | No | Rat study Mouse study | Intra-myocardial injection; miR-22 loss-of-function genetic model in the mouse | KDM3A gene knockout rats; miR-22 loss-of-function genetic model in the mouse | miR-22-3p depletion activates cardiac autophagy, prevents post-MI remodeling, and reduces the infarct area. Loss of miR-22 sensitized mice to the development of dilated cardiomyopathy | [136] [137] |
| miR-25-3p | Inhibition of miR-25 ameliorates cardiac function | No | No | Mouse study | i.v. | Mouse heart failure model | Anti-miR-25 enhances cardiac contractility and function through SERCA2a restoration | [138] |
| miR-34a-5p | miR-34a is a naturally occurring tumor suppressor that is underexpressed in a broad range of tumor types miR-34a deficiency improves survival of cardiomyocytes and VSMCs, revascularization, and prevents vascular calcification | Yes No | A liposomal double-stranded miR-34a mimic (MRX34) No | Phase I (NCT01829971) 85 patients (terminated) Rat study | iv., different doses (70/90/110mg/m2) and various schemes i.v. | Adult patients with various solid tumors refractory to standard treatments Evaluation of miR-34a-5p knockdown on cardiac apoptosis and I/R injury after MI | Terminated due to severe immune-mediated toxicities, resulting in four deaths related to miR-34a injection The delivery of anti-miR-34a molecules is effective to treat vascular disease, but considering the tumor suppressor activity of miR-34a, systemic delivery could promote tumorigenesis | [124] [125] |
| miR-92a | AntagomiR-92a reduces infarct size | No | No | Mouse, pig models | intracardiac | Model of acute MI | Reduced cardiac ischemia and post-MI remodeling, enhanced blood vessel growth and functional recovery of damaged tissues | [119, 120] |
| miR-126-5p | The 5p strand sustains endothelial integrity in the context of high shear stress and autophagy | Yes | No | Peripheral blood cells co-transfected with miR-126-5p mimic | Peripheral blood samples | 30 patients with atherosclerosis | Prevented atherosclerosis, suppressed proliferation and migration of ox-LDL-VSMCs through upregulating miR-126-5p | [139] [140] |
| miR-148a-3p | Elevation of miR-148a preserves cardiac function | Yes | No | Mouse study | i.v. | Heart failure TAC surgery mouse model | Elevation of miR-148a expression protects the myocardium from dilation and cardiac dysfunction | [141] |
| miR-199-5p | Inhibition of miR-199b prevents cardiac remodeling | No | No | Mouse study | Cell culture of ECs of the heart | STAT3-KO spontaneous heart failure mouse model | Protective effects of downregulated miR-199a-5p on ischemic and hypoxic cardiomyocytes | [142] |
| miR-208a-3p | AntimiR prevents cardiac remodeling, preserves cardiac function | No | No | Rat and pig study | s.c. | Rat MI model and porcine ischemia-reperfusion model | Level of stress influences the level of target depression after anti-miR-208a treatment | [143] |
| miR-214-3p | miR-214-3p is an important repressor for fibroblast proliferation and fibroblast-to-myofibroblast transition | yes | No | Mouse study | i.v. | TAC-induced murine model | miR-214 deficiency impairs cardiomyocyte survival, increases cardiac fibrosis | [144] |
| miR-221/222-3p | miR-222 elevation protects from cardiac remodeling | No | No | Rat study | Intra-carotid infusion | Carotid injury rat model | Re-endothelialization was increased by miR-221/222 inhibition | [145] [146] |
[i] ASO – antisense oligonucleotide, EVs – extracellular vesicles, ICM – ischemic cardiomyopathy, i.v. – intravenously, LNA – locked nucleic acid modified, LVEF – left ventricular ejection fraction, NAFLD – non-alcoholic fatty liver disease, NASH – non-alcoholic steatohepatitis, NT-proBNP – amino terminal fragment of pro-brain natriuretic peptide.
In line with this observation, preclinical data encourage the use of miR-590-3p and miRNA-199a-3p, which regulate calcium signaling through HOMER1 [121]. They possess pro-proliferative effects in cardiomyocytes through DNA synthesis and increased cytokinesis. They are also involved in reducing fibrotic scar size and can improve cardiac function following MI in neonatal mice [121]. In addition, the miR-302-367 cluster can induce and maintain pluripotency, leading to an increase in cardiomyocyte mass, decreased myocardial fibrosis, and improved function in a failing myocardium [122]. In the preclinical stage, there are studies on the use of miR-29 mimics for the treatment of cardiac fibrosis, and studies on inhibitors of miR-15 [111]. Also, anti-miR-212 and anti-miR-652 demonstrated a good anti-fibrotic effect [116].
However, not all candidate microRNAs for therapy were so successful (Table IV) [123–146]. On contrary, there are more microRNAs that failed than those that succeeded. Clinical studies with anti-miR-103/107-3p (AZD4076), miR-34a mimics, and anti-miR-155-5p (cobomarsen) were terminated or halted by the sponsor for strategic reasons (severe immune-related side-effects) [110, 111, 123–125], although in preclinical studies they could be considered for therapy in cardiovascular disease [124, 125].
Confusing data regard studies with miR-21 antagonists and miR-21 mimics [126]. miR-21 function depends on its source, as it derives from diverse cell types, including ECs, platelets, VSMCs, cardiomyocytes, and fibroblasts. miR-21 participates in infarction injuries, cardiac remodeling, atherosclerosis, arrhythmias, and cardiomyopathy [126]. However, increased miR-21 expression caused by MI protected cardiomyocytes from apoptosis (positive action), along with enhancing the activation of cardiac fibroblasts (negative action) [126]. Furthermore, miR-21-targeted PPARa resulted in increased inflammation owing to the ECs in the heart. Eventually, miR-21 therapy showed many off-target effects in the liver, lung, and kidney with a concomitant increase in serum creatinine levels [126].
Regenerative cardiology – reprogramming heart cells. Prevention of cardiomyocytes loss
During cardiac injury (e.g., resulting from MI), large numbers of cardiomyocytes are lost due to apoptosis, autophagy, or necrosis [14, 147–152]. The accompanying recruitment of monocytes results in the removal of any damaged/necrotic cardiomyocytes. This process results in cardiac muscle reduction, leading to HF, which is associated with malignant arrhythmia and increased mortality risk.
One approach is to inhibit the extensive apoptosis of cardiomyocytes, which can be accomplished via decreasing the expression of pro-apoptotic microRNAs (Table V). Among these, anti-miR-195 was shown to regulate anti-apoptotic genes; however, it is unclear whether miR-195 antagonists can reverse cell senescence or the apoptotic rate [153]. miR-199a was shown to directly target Clic5, which is involved in the proliferation of adult cardiomyocytes [143, 150, 151]. Although miR-199a has been reported to improve cardiac function, its prolonged administration led to lethal cardiac arrest [150, 151]. Finally, miR-125b overexpression efficiently attenuated cardiac function injury of HF mice by targeting BAK1 through inhibiting cardiomyocyte apoptosis, suggesting that the miR-125b/BAK1 axis might be a potential target for the diagnosis or treatment of HF [152].
Table V
MicroRNA-based therapies in preclinical studies used for cardiomyocytes protection and proliferation
| microRNA | Studied therapeutic agent | Subject to study | Activity/significance/side-effects | Reference |
|---|---|---|---|---|
| miR-195 | Anti-miR-195 regulates anti-apoptotic genes, can decrease apoptosis after ACS | In vitro cultured myoblast | miR-195 inhibition did not affect cell ageing or rejuvenation of human skeletal muscle-derived stem/progenitor cells (SkMDS/PCs). It is unclear whether miR-195 antagonists can reverse cell senescence or the apoptotic rate | [153] |
| miR-199a | miR-199a was shown to directly target Clic5, which is involved in the proliferation of adult cardiomyocytes. | Mice | Although miR-199a has been reported to improve cardiac function, its prolonged administration led to lethal cardiac arrest | [143, 150] |
| miR-125b | miR-125b agonists | Mice | miR-125b overexpression efficiently attenuated cardiac function injury of heart failure mice by targeting BAK1 through inhibiting cardiomyocyte apoptosis, suggesting that the miR-125b/BAK1 axis might be a potential target for the diagnosis or treatment of HF | [152] |
| A cocktail of 4 microRNAs mimics (miR combo) miR-1, miR-133, miR-208, and miR-499 | May be efficient in the induction of transformation of fibroblasts into induced cardiomyocytes (iCMs) | Adult human cardiac fibroblasts | The percentage of cTn-positive cells (former fibroblasts) 15 days after miR combo transfection was ∼11%, as evaluated by flow cytometry. A potential new strategy for myocardial regeneration after ACS | [114] |
| A cocktail of 4 microRNAs mimics (miR combo) miR-1, miR-133, miR-208, and miR-499 | miR combo effectively reprograms fibroblasts of any mammalian species into cardiomyocytes | Cardiac fibroblasts of pig, dog and fetal human cardiac fibroblasts | miR combo reprograms pig, dog, and fetal human cardiac fibroblasts into cardiomyocyte-like cells that induce the expression of sarcomere and cardiac ion channels. In a fetal human cardiac fibroblast model ∼10% of cells had cardiomyocyte-like properties 14 days after transfection. This study validates the miR combo as a potential therapeutic modality for myocardial regeneration following ACS | [158] |
Another approach is the reprogramming of fibroblasts into induced cardiac progenitor cells (iCPCs) to obtain de novo cardiac lineages [108, 154–160]. Cardiomyocyte-derived microRNAs such as miR-1 (through the target gene Bcl2), miR-133a (many target genes), miR-208a (APC), miR-223 (PARP-1), miR-499, and miR-145 (FRS2) may play important roles in these processes [115, 150].
A cocktail of 4 microRNAs called the miR combo (including mimics of miR-1, miR-133, miR-208, and miR-499) is proposed to be efficient in the induction of fibroblast transformation into induced cardiomyocytes (iCMs) (Table V) [154]. The researchers have also developed a delivery system to introduce the miR combo directly into the heart [155]. Yang et al. delivered a cocktail of 4 cardiomyocyte-derived microRNA mimics using nanocomplexes of branched polyethyleneimine-coated nitrogen-enriched carbon dots, which were then injected into the heart [156]. These nanocomplexes led to the efficient direct reprogramming of fibroblasts into iCMs without genomic integration and resulted in effective recovery of cardiac function following MI [156, 157]. MicroRNAs have been used to enhance direct reprogramming alone or in combination with transcription factors. Studies by Jayawardena et al. and Jiang et al. reported that a transient non-viral transfection system of miR-combo including miR-1, miR-133, miR-208, and miR-499a was able to reprogram mouse cardiac fibroblasts into iCMs in vitro and in vivo [157]. Recently, Baksh et al. demonstrated that miR combo reprograms pig, dog, and fetal human cardiac fibroblasts into cardiomyocyte-like cells that induce the expression of sarcomere and cardiac ion channels [158]. In a fetal human cardiac fibroblast model, approximately 10% of cells had cardiomyocyte-like properties 14 days after transfection. This study is important as it validates the miR combo as a potential therapeutic modality for myocardial regeneration following ACS [158].
Combining microRNA-based therapies with other emerging therapies, such as cardiac stem cell or gene therapies.
Treating cardiovascular diseases with cardiac stem cells is a valid treatment. Cardiac stem cells can reproduce the myocardial cells [66, 161, 162]. Studies have shown that cardiac stem cell proliferation and differentiation are regulated by microRNAs [161, 162]. How microRNAs regulate cardiac stem cell behavior is an interesting area of research that can help us study and control the function of these cells in vitro – an achievement that may be beneficial for patients with cardiovascular diseases [161, 162].
Limitations of microRNA use.
It is important to understand the limitations of microRNAs. Many microRNAs can be used as diagnostic and prognostic markers without causing harm. However, even though microRNA mimics and antagonists are already being investigated in animal models, or even in human, their main limitations concern their therapeutic safety, stability in circulation, toxicity, unwanted off-target actions, and lack of therapeutic actions in humans that were previously described in cultures or animal models [110, 111]. Important issues to understand are their potential toxicity, off-target actions, and irreversible inhibition of mRNA. Some may act as oncogenes or exert dual/multiple roles, including opposite actions. Because of this, delivery systems were developed to administer microRNAs locally (e.g., directly into the coronary circulation or cardiac muscle) [110, 111]. To prevent their quick degradation in the blood stream, synthetic oligonucleotides that are more resistant to enzymatic lysis were produced.
Future directions and conclusions
Two approaches for the management of atherosclerosis can be considered for testing in the clinical setting. The first one, presented in this review, is associated with a reduction in atherosclerosis-related post-ischemic complications [108–110]. This approach attempts to inhibit fibroblast proliferation and scarring, harmful apoptosis, autophagy or necrosis of cardiomyocytes, and involves the reprogramming of fibroblasts into iCMs, which may offer better post-ACS recovery and a decreased rate of post-MI adverse events.
The second approach involves early intervention at the level of atherosclerotic growth initiation [4, 5, 74]. The optimal point for early intervention would be during the identification of intima-media complex thickening or the presence of early plaques [127–129]. Increasing thickness of the carotid intima-media complex and carotid plaques is related to the extent and severity of atherosclerosis, in addition to polyvascular atherosclerosis [163–171]. Also, the role of microRNAs in cardiovascular risk factor modification seems extremely important. microRNAs are highly expressed in patients with obesity, metabolic syndrome and familial hyperlipidemia [172, 173]. Particularly miR-150 showed associations with proprotein convertase subtilisin/kexin type 9 (PCSK9) [171]. PCSK9 regulates extracellular vesicle-derived microRNAs, especially those involved in inflammation and expression of the low-density lipoprotein receptor (LDLR) receptor. Studies on mice demonstrated a downregulation effect of immunotherapy with the PCSK9 peptide vaccine on the hepatic expression levels of miR-27a [174]. miR-27a has been shown to improve LDLR degradation by directly binding to its 3′-untranslated region (UTR) and indirectly by enhancing PCSK9 [174]. Therefore, respective microRNAs might be significantly regulated by statin therapy [175–177]. As mentioned before, also treatment with angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, GLP-1, PCSK9 inhibitors, and antiplatelets modulates expression levels of corresponding microRNAs [175–177].
Cellular and molecular biology is a rapidly growing field of research [178, 179]. Soon, it may provide novel solutions for personalized treatment, integrating knowledge from basic cardiological science to preclinical studies, and eventually to clinical practice. The clinical approaches are worthy of continued research and development. In both treatment strategies, microRNAs may play a critical role in the regeneration of the heart, as well as in the initiation and progression of atherosclerosis. While initial preclinical data are promising, there is still much work to be done in this field.