Introduction
Severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2), the etiologic agent of the current outbreak of coronavirus disease 2019 (COVID-19), presents great similarity to the other coronaviruses SARS-CoV-1 and MERS-CoV, which caused the severe acute respiratory syndrome (SARS in 2002–2003) and Middle East respiratory syndrome (MERS from 2012-ongoing) outbreaks, respectively. These are enveloped viruses and share high homology in their structures [1].
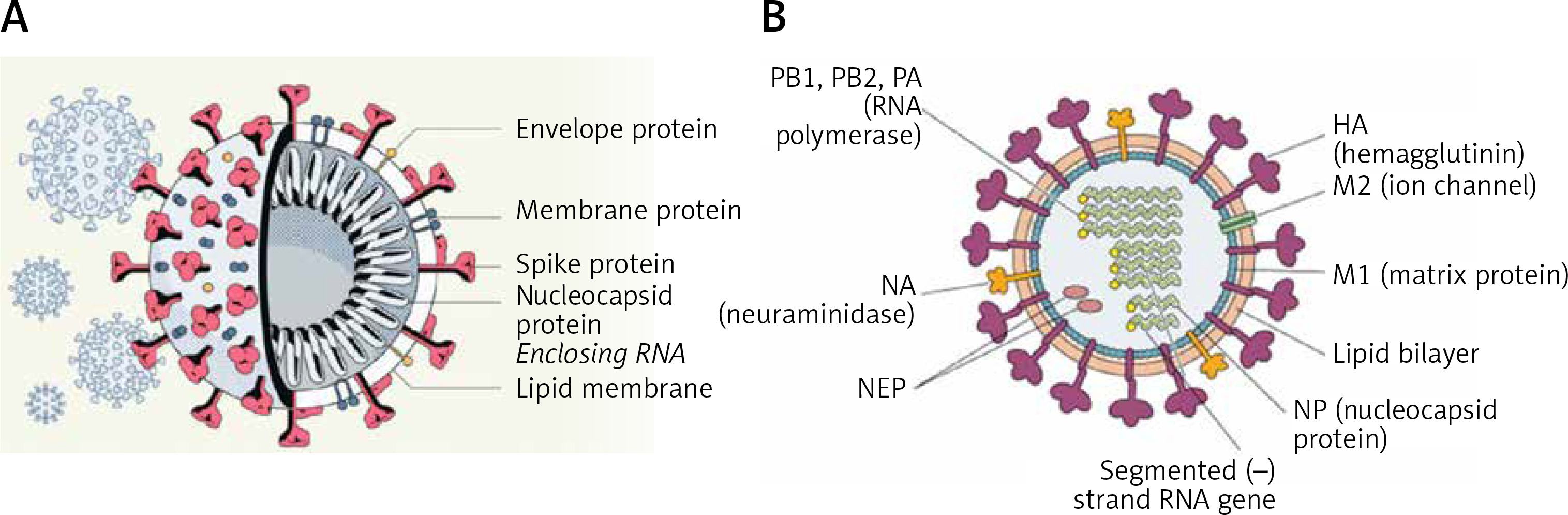
SARS-CoV-2 virus particles contain proteins known as spikes protruding from their surface. These spike proteins bind to human cells to enable the viral membrane to fuse with the cell membrane that facilitate the viral genes to enter the host cell to produce its actions (see Figure 1 for structure of SARS-CoV-2). Even though the sequences and structures of the spikes of SARS-CoV-2 and SARS-CoV-1 are similar, antibodies directed against SARS-CoV-1 do not bind to the SARS-CoV-2 spike protein [2, 3] though some amount of limited cross reactivity has bene reported [4–6]. This indicates that more specific antibodies need to be generated for their potential use in humans.
Figure 1
A – Structure of SARS-CoV-2 that causes COVID-19. Note the structural similarity with swine flu virus given in Figure 1 B. B – Structure of the swine flu A/Mexico/09 (H1N1) virus. For comparison between influenza virus and coronavirus see Table II
Current strategies that are being employed against COVID-19 include inhibitors of viral entry into cells, viral replication, and RNA synthesis. SARS-CoV-2 utilizes angiotensin-converting enzyme 2 (ACE2) and the cellular protease transmembrane protease serine 2 (TMPRSS2) to enter target cells. Hence, chemicals that act on these two ligands are under development. But these strategies take considerable time to come to the clinic. SARS-CoV-2 spike glycoprotein (S protein) is cleaved into S1 and S2 subunits at the time of infection. S1 contains the receptor binding domain (RBD) that binds to the peptidase domain (PD) of ACE2, while S2 is needed for membrane fusion. When S1 binds to ACE2, another cleavage site on S2 is exposed that is cleaved by host proteases to facilitate viral infection. It is noteworthy that the S protein of SARS-CoV-2 exploits ACE2 for host infection [7]. ACE2 is expressed in many tissues including but not limited to lungs, heart, kidney, and intestines, which are the targets of SARS-CoV-2. ACE2 is needed for the maturation of angiotensin to control vasoconstriction and blood pressure. Reduced expression of ACE2 is associated with cardiovascular diseases, which explains why many with COVID-19 may develop myocardia damage and heart failure (see Figure 1 for the structure of SARS-CoV-2; and similarities and differences between Swine flu A/Mexico/09 (H1N1) virus and SARS-CoV-2; Table I for similarities and differences between influenza and coronaviruses and Figure 2 for the role of ACE2 in humans).
Table I
Comparison between influenza virus and coronavirus (see Figures 1 A and B also)
Table II
Clinical features of COVID-19
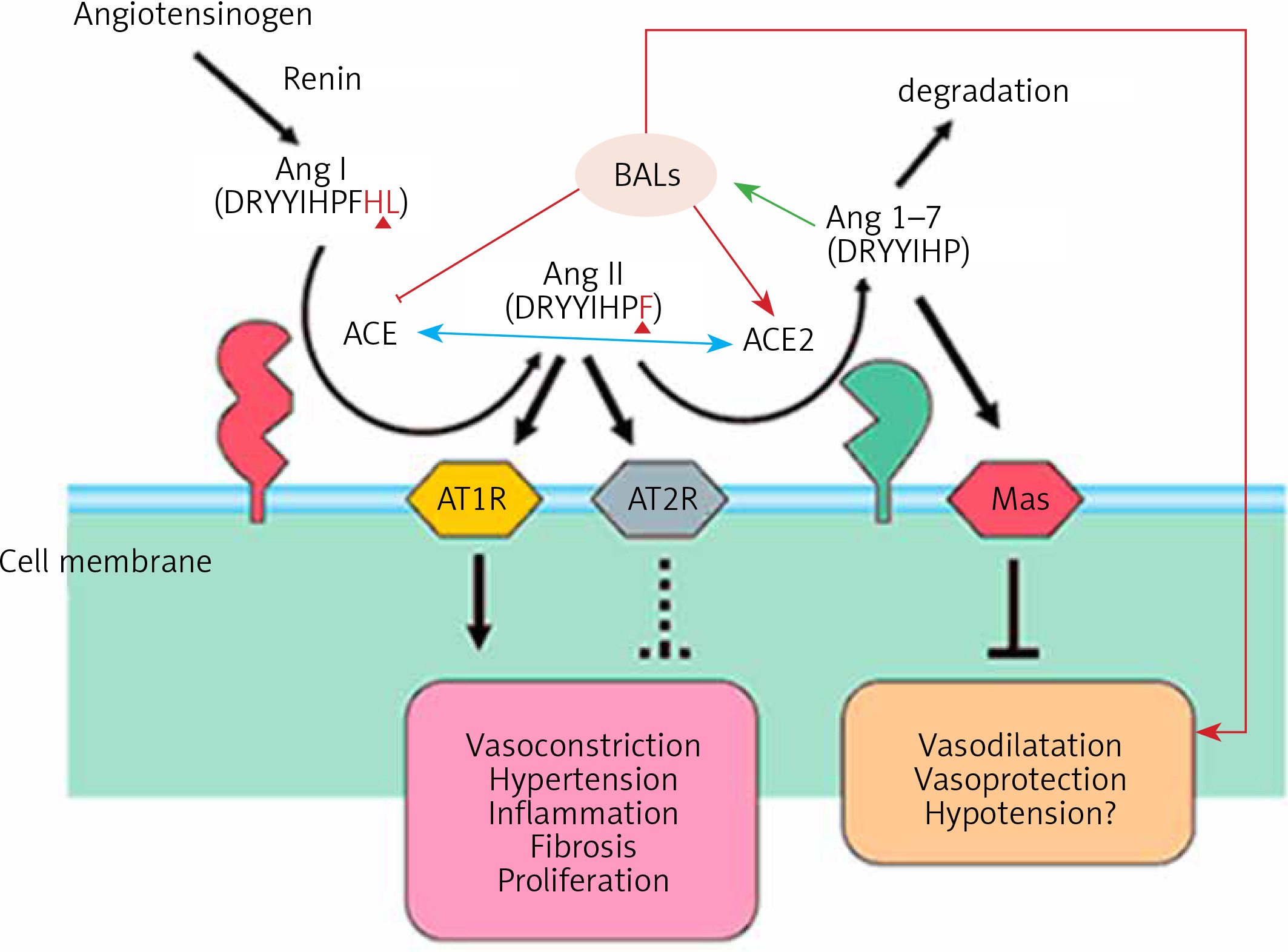
Figure 2
Scheme showing the actions of ACE and ACE2 and their role in the regulation of blood pressure, humoral balance, inflammation, cell proliferation, hypertrophy, and fibrosis. The ACE/Ang /AT1R axis and the ACE2/Ang 1–7/MAS axis balance each other. Ang 1–7 is known to restore polyunsaturated fatty acid content of cells to normal especially in diabetes mellitus. Thus implies that there is a close interaction between Ang 1–7 and BALs and possibly some of the beneficial actions of Ang 1–7 on vascular tissue could be due to its actions on EFA metabolism (see Singh K, Singh T, Sharma PL. J Exp Pharmacol 2010: 2: 163-8. DOI: 10.2147/JEP.S14342)
Historical aspects of influenza
Influenza, or flu, attacks principally the respiratory system and is highly contagious. The infection can spread from one person to another through droplets generated while coughing, sneezing or talking. These droplets can travel into the air and eventually be inhaled by others, leading to spread of the disease. In addition, the virus can spread by touch when one person touches any infected surface with the virus and then touches his or her mouth, eyes, or nose. In the USA, during the flu season more than 200,000 are hospitalized for flu-related complications. Young children, the elderly (> 65 years), pregnant women and people with asthma, diabetes mellitus and/or heart disease are particularly vulnerable to the risk of flu-related complications, including but not limited to pneumonia, myocarditis, central nervous system (CNS), eye, ear and sinus infections and bronchitis. Recent studies suggested that SARS-CoV-2 can infect neuronal cells, which can result in an encephalitis-like clinical presentation, produce thrombosis of the cerebral veins and arteries leading to stroke and other complications. It needs to be noted that post-COVID-19 manifestations include persistent headache, memory loss, confusion, inability to concentrate, breathlessness, cough, low grade fever, muscle pains and unexplained weakness [8–13].
There have been several influenza pandemics. The first flu pandemic was recorded in 1918 and termed “Spanish flu” (this is a misnomer as it did not originate in Spain and is more aptly termed the “1918 influenza pandemic,” or the “1918 flu pandemic,”) in which the death toll is estimated at ~20 million to 50 million worldwide. The flu pandemic of 1957–1958 killed ~2 million people worldwide. The pandemic of 1968-1969 killed ~1 million, whereas > 12,000 Americans died during the H1N1 (or “swine flu”) pandemic of 2009–2010. The current pandemic of COVID-19 is spreading rapidly across all nations and its final impact is yet to be gauged as the pandemic is still continuing. The current COVID-19 pandemic is particularly deadly since many carriers are asymptomatic. It is estimated that ~40% of those infected with SARS-CoV-2 are asymptomatic and are responsible for spreading the infection.
In general, most influenza outbreaks seem to be disproportionately lethal to the very young and elderly. The surprisingly higher than expected mortality during the flu pandemic of 1918 in young adults has been attributed to the triggering of a cytokine storm. Another explanation offered for the higher mortality in the young during this pandemic is the superadded bacterial infection [14–16]. It is rather paradoxical to think that the strong immune reaction is harmful, whereas the limited immune response of children and middle-aged adults is much less lethal.
Structure of influenza virus
The structure of the influenza virion is similar to SARS-CoV-2 and both are enveloped viruses (Figure 2). It is important to understand the similarities and differences in the structure of influenza and SARS-CoV-2 viruses as shown in Table I. These differences may underlie the variations seen between influenza-induced illness and SARS-CoV-2-induced severe illness in the form of ARDS (acute respiratory distress syndrome), cytokine storm and thrombosis of the lung capillaries and vessels, stroke, myocarditis and other features.
COVID-19 and ACE2
Even though COVID-19 is primarily a respiratory infection, it is now known that its manifestations may be seen in almost all systems of the body including cardiovascular and immune systems. Those who have pre-existing cardiovascular conditions are particularly vulnerable to disproportionately worse outcomes, with almost 5–10-fold higher mortality [17, 18].
There are many similarities between SARS-CoV-2 and SARS-CoV. SARs-CoV-2 and SARS-CoV spike proteins share 76.5% identity in amino acid sequences and their spike proteins have a high degree of homology [19–22]. It has been shown that residue 394 (glutamine) in the SARS-CoV-2 receptor-binding domain (RBD), corresponding to residue 479 in SARS-CoV, is recognized by the critical lysine 31 on the human ACE2 receptor [23]. SARS-CoV-2 recognizes and binds to human ACE2 more avidly compared to SARS-CoV [24–26]. It is known that blocking the renin-angiotensin pathway attenuates or at least reduces the severity of the lung damage due to SARS-CoV-2. This suggests that ACE2 not only is needed for the virus to enter the target cell but also protects from lung injury.
ACE and its homologue ACE2 regulate the renin–angiotensin system. ACE cleaves the decapeptide angiotensin (Ang)I into the octapeptide AngII, whereas ACE2 cleaves a single residue from AngI to generate Ang1–9 and a residue from AngII to generate Ang1–7. Thus, ACE2 exerts negative regulatory control on the renin–angiotensin system by inactivating AngII (Figure 2). It is known that in ARDS (acute respiratory distress syndrome) there is marked downregulation of the ACE2 protein while ACE levels remain constant. ARDS can be improved by catalytically active ACE2. AngII levels in the lungs and plasma are markedly increased in ARDS with a concomitant decrease of ACE2 expression. Thus, ACE promotes ARDS through increased AngII production whereas ACE2 protects against ARDS [27]. In other words, inhibition of ACE activity may lead to a decrease in the activity and receptor number of ACE-2 due to the feedback regulation that exists between ACE and ACE-2. The host cell entry of SARS-CoV-2 depends on the ACE2 receptor and can be blocked by inhibiting the cellular serine protease TMPRSS2 that is employed by SARS-CoV-2 for S protein priming.
Clinical picture of COVID-19
The most prominent and typical symptoms of COVID-19 are cough, fever, muscle aches, headaches and difficulty breathing that may lead to the development of ARDS, necessitating some of these patients to be put on ventilator and extracorporeal membrane oxygenation (ECMO). It is estimated that about 20–30% of the patients put on ventilators eventually succumb to the disease [28–30]. In addition, some patients may have headache, dizziness, abdominal pain, diarrhea, nausea, and vomiting. With the onset of alveolar damage, these patients will have progressive respiratory failure that may lead to death (see Table II for common clinical features of COVID-19).
Since ACE2 is needed for host cell entry and subsequent viral replication, efforts have been made to study its expression and distribution in the human body to determine the potential infection routes of SARS-CoV-2. High ACE2 expression is seen in type II alveolar cells (AT2) of lung, esophagus upper and stratified epithelial cells, enterocytes from the ileum and colon, cholangiocytes, myocardial cells, kidney proximal tubule cells, and bladder urothelial cells [31–33]. The ACE2 protein is present in various human organs including oral and nasal mucosa, nasopharynx, lung, stomach, small intestine, colon, skin, lymph nodes, thymus, bone marrow, spleen, liver, kidney, and brain. Expression of the ACE2 protein on lung alveolar epithelial cells and enterocytes of the small intestine, vascular endothelial cells, and arterial smooth muscle cells of several organs may account for damage to these tissues during COVID-19. This, in turn, may lead to the development of ARDS, thromboembolic manifestations and gastrointestinal disorders. The wide distribution of ACE2 in several human organs provides a clue as to the COVID-19 disease manifestations especially regarding the vascular complications such as thrombosis seen in peripheral blood vessels and in the smaller blood vessels of lungs.
Studies have invesigated the localization of ACE2 protein in various human organs (oral and nasal mucosa, nasopharynx, lung, stomach, small intestine, colon, skin, lymph nodes, thymus, bone marrow, spleen, liver, kidney, and brain). The most remarkable finding was the surface expression of ACE2 protein on lung alveolar epithelial cells and enterocytes of the small intestine. Furthermore, ACE2 is present in arterial and venous endothelial cells and arterial smooth muscle cells in all organs studied. In conclusion, ACE2 is abundantly present in humans in the epithelia of the lung and small intestine, which might provide possible routes of entry for the SARS-CoV. This epithelial expression, together with the presence of ACE2 in vascular endothelium, also provides a first step in understanding the pathogenesis of the main SARS disease manifestations.
Since several tissues contain high ACE2-expressing cells, they are potential sources through which SARS-CoV-2 can enter the body to produce its infection. In addition to these cells/tissues, another potential source of entry for the SARS-CoV-2 virus is the mucosa of the oral cavity, which has been shown to express ACE2 and is highly enriched in epithelial cells. Among different oral sites, ACE2 expression is higher in tongue than buccal and gingival tissues [33]. This observation lends support to the observation that SARS-CoV-2 can be shed by oral droplets and saliva and mucosa of the oral cavity is a potential route of infection. Since many organs/tissues have the ACE2 receptor and SARS-CoV-2 is able to infect almost all tissues in the body, it is likely that SARS-CoV-2 can be found in the saliva, urine, feces, and other secretions. This is supported by the recent report that SARS-CoV-2 has been found in the sewage of several cities [34–37]. Hence, it is imperative that efforts be made to determine whether SARS-CoV-2 is present in all the urine, feces, and saliva of those suspected to have the disease. It is important to know the source and route of infection to take suitable preventive measures to stop the spread of COVID-19. It is important to note that SARS-CoV-2 has been detected in the municipal water supply and in sewage water, a possible source of infection. This assumes significance especially for third world countries (such as India) where the sewage and drinking water supply systems are not adequate. Since housing is inadequate and more than 5–6 persons live under one roof and washroom facilities are limited, the possibility of water source spread of COVID-19 is high. This coupled with the lack of adequate facilities for testing (both RT-PR and antibody) further aggravates the spread of COVID-19 in these countries. This is supported by the belief that in India with a population of almost 1.3 billion for one detected COVID-19 case there could be almost 300–400 undetected cases, implying that there could be almost 300 million infected with SARS-CoV-2.
Some of the unusual manifestations of COVID-19 include anosmia and dysgeusia, skin rashes, diarrhea, kidney abnormalities and potentially life-threatening blood clots.
Anosmia (loss of sense of smell) and dysgeusia (alteration of sense of taste) are the most common early manifestation of COVID-19 that may go unnoticed, especially in the elderly. It was reported that anosmia could be present in ~73% of patients long before COVID-19 diagnosis. Improvement in anosmia may occur in ~27% of patients. The mean time to improvement was reported to be ~7.2 days, whereas 85% showed improvement within 10 days [38]. These observations indicate that anosmia can be a presenting symptom of COVID-19. Anosmia may be critical to identify individuals infected with SARS-CoV2.
Skin rashes and COVID toes
Similar to many viral infections such as chicken pox or herpes, patients with COVID-19 also may show skin rashes that could be due to either direct invasion of the skin by the virus or immune reaction to the virus as seen in Epstein-Barr virus or West Nile infections. Some COVID-19 patients, especially children, show head-to-toe red rashes, hive-like eruptions, blister-like bubbles and even lacy, purplish rashes spreading across larger patches of skin, and some have red, tender bumps that appear around the toes and heels–dubbed “COVID toes”. It is suspected that those with lacy purple rashes are more likely to develop blood clots. Though it is not clear whether SARS-CoV-2 itself can cause rashes, it is likely that the manifestations are a result of an inert particle reaction in the skin, initiating an inflammatory process in response to circulating antibodies and sensitized lymphocytes. Histological examination of the skin biopsy of these rashes showed that the findings are very similar to hand-foot-and-mouth disease and the RT-PCR for SARS-CoV-2 from the vesicles was negative. This negative reaction could be due to low viral load or absence of the virus itself. This suggests that contact with the skin rashes may not be infective [39].
Gut manifestations
Since intestinal epithelial cells are rich in ACE2, it is no wonder that the gut is also a major target of SARS-CoV-2. Some of the gastrointestinal manifestations of COVID-19 include diarrhea, nausea and vomiting. It appears that those who have gastrointestinal symptoms tend to be diagnosed later and endure longer infections (partly because they are misdiagnosed with the mistaken belief that gut symptoms are unlikely due to COVID-19). It needs to be emphasized that the gut is a massive immune organ and so when it is infected it takes a long time to clear the virus from such a large organ. Since the gut can be involved in COVID-19, it stands to reason that SARS-CoV-2 can be excreted in feces, which also could constitute a source of infection. Under normal conditions, the acid in the stomach is expected to inactivate the virus. Hence, use of antacids or proton pump inhibitors that suppress acid production may create a fertile ground for SARS-CoV-2 to proliferate and travel to the other parts of the gut. In contrast, it was reported that H2 blockers could be of benefit in COVID-19 [40, 41]. It has been proposed that SARS-CoV-2 may induce differentiation and activate mast cells. Such activated mast cells release histamine, proteases, cytokines, chemokines, and arachidonic acid derived prostaglandins (PGs) and leukotrienes (LTs) that have a role in inflammation. Histamine is known to be involved in the expression of chemokine IL-8 and cytokine IL-6, an effect that can be inhibited by H2 blockers. Alveolar macrophages activated by SARS-CoV-2 release IL-1 that, in turn, stimulates mast cells to produce IL-6. Both IL-1 and IL-6 can work together to produce excessive inflammation as seen in COVID-19. Furthermore, histamine enhances IL-1-induced IL-6 gene expression and protein synthesis via H2 receptors in peripheral monocytes. Thus, histamine released by mast cells during an inflammatory reaction as seen in COVID-19 can increase production of IL-1 to amplify the inflammatory process in the lung infected with SARS-CoV-2 [40]. This work was supported by in silico work that showed that famotidine could interact with the catalytic site of the three proteases associated with SARS-CoV-2 and suggested that its intravenous administration could be of significant benefit in COVID-19 [41]. This proposal is supported by the observation that famotidine use was associated with a better outcome in those with COVID-19 [42–44]. These initial observations need to be confirmed in large-scale double-blind placebo controlled clinical trials.
Renovascular system
The function of ACE in the kidney is to convert Ang I to Ang II to regulate blood pressure and salt and water balance (through aldosterone secretion modulation). Pulmonary epithelium is the main source of ACE. Intrarenal ACE production in the human kidney is about five times higher compared to ACE found in the human lung [45, 46]. A remarkably high concentration of ACE is present in the brush border of the proximal tubule, glomerular endothelium, mesangial cells, podocytes, and distal nephron [47–51]. The purpose of the ACE2 enzyme is to counteract many functions of ACE (Figure 2). ACE2 is needed to convert Ang II into the vasodilator and antiproliferative Ang 1–7 and Ang I into the inactive Ang 1–9. Ang 1–7 is an antioxidant, anti-fibrotic, and anti-inflammatory molecule, and acts through its receptor Mas (MasR) [52, 53]. ACE2 expression is also seen in the heart, liver, lung, and neurons [54–56]. ACE2 expression is present in significant amounts in tubular and glomerular epithelium, vascular smooth muscle cells, the endothelium of interlobular arteries, and glomerular mesangial cells [57, 58]. SARS-CoV-2 infection is seen in the kidney due to the presence of ACE2. SARS-CoV-2 can be detected in the urine of infected people. Acute kidney injury among COVID-19 patients is seen in almost 36.6% of patients admitted and some of them need dialysis [59, 60]. More importantly, occurrence of renal injury due to COVID-19 could be correlated with severity of the respiratory symptoms. It is likely that SARS-CoV-2 can directly affect the kidneys and the resultant inflammatory response may worsen not only the respiratory involvement but also if there is any previous renal disease. The cytokines that are released as a result of SARS-CoV-2 infection can affect the blood vessels, leading to renal ischemia that may worsen renal function, necessitating appropriate interventions in the form of dialysis. This suggests that all those who have been diagnosed with COVID-19 need to be assessed for their renal status and checked for hematuria and urinary albumin presence, an indication of renal damage. But it is not yet clear whether COVID-19 can cause any long-lasting renal damage and, if so, what that could be.
Hepatobiliary system
Hepatocytes have the ACE2 receptor and so are also a target of SARS-CoV-2 infection. Further, more than half of the patients admitted for COVID-19 show alterations (higher or lower) in the levels of liver enzymes, indicating that the liver has been affected. But it is apparent that liver cell failure is unlikely and so hepatic functions are likely to revert to normal over a period of time. The alterations seen in hepatic enzymes in COVID-19 could be due to either direct infection of hepatocytes by SARS-CoV-2 or secondary to the inflammatory response seen in these patients. Lymphocytic infiltration associated with sinusoidal dilatation, steatosis, and multifocal hepatic necrosis can be seen in COVID-19 [61]. In the event these patients have pre-existing liver disease, it is likely to be exaggerated and may lead to further damage to liver in view of the inflammatory events associated with COVID-19.
Thrombosis and its consequences in COVID-19
Vascular endothelial cells carry the ACE2 receptor, which implies that SARS-CoV-2 can directly attack the blood vessels, especially of the lungs, resulting in thrombosis in the pulmonary blood vessels (both venous and arterial) due to excessive inflammation, platelet activation, endothelial dysfunction, and stasis that contributes to ARDS seen in these patients. Some patients with COVID-19, especially those who have severe disease, may have vascular thrombosis (due to endothelialitis) in the legs and lungs and sometimes these patients develop disseminated intravascular coagulation. This led to the suggestion that anti-thrombotic therapy may be of significant benefit to these patients.
These presumptions are supported by a recent study which showed that those who died from COVID-19 or influenza-associated respiratory failure showed diffuse alveolar damage with perivascular T-cell infiltration. COVID-19 specimens also showed severe endothelial injury with intracellular virus and disrupted cell membranes, whereas the pulmonary vessels revealed extensive thrombosis with microangiopathy and alveolar capillary microthrombi with an extremely high prevalence of intussusceptive angiogenesis compared to those with influenza. These results suggest that vascular angiogenesis pulmonary pathology of COVID-19 is distinctly different from that of equally severe influenza infection [62].
It has been reported that remarkably high numbers of ACE2-positive cells are present in the lungs of those with COVID-19 compared to uninfected controls. Vascular endothelial cells are the main target of SARS-CoV-2, which can lead to disruption of intercellular junctions, cell swelling, and a loss of contact with the basal membrane. SARS-CoV-2 virus has been demonstrated within the endothelial cells, suggesting that the virus has a direct effect on these cells (endothelial cells) that may be exaggerated by the perivascular inflammation noted [62]. Since many COVID-19 patients recover completely even after ventilatory therapy, it can be assumed that several features described [62] are reversible though the time needed for this reversal to normal remains to be determined. It is also equally true that we still do not know whether all those who have recovered from COVID-19 have any residual pulmonary damage and if so, what their long-term consequences are. Nevertheless, the presence of extensive capillary microthrombi in those with COVID-19 suggests that early institution of anti-coagulant therapy is warranted. Thrombosis of cerebral vessels leading to stroke is more common in young people, suggesting that stroke in the young needs to be assessed for the presence of SARS-CoV-2 infection. The big question is, do many, if not all, cases of stroke in the young need to be investigated for SARS-CoV-2 even during a non-epidemic or non-pandemic period? One investigation that may help is the measurement of D-dimer in these patients. Similarly, coronary heart disease in the young with no known risk factors also needs to be investigated for the presence of silent COVID-19 infection. This aspect needs special attention as ~80% of patients with SARS-CoV-2 infection are asymptomatic.
Another peculiar manifestation seen in COVID-19 is the complete lack of clot lysis at 30 min on a thromboelastogram (TEG) assay coupled with a D-dimer value above 2600 ng/ml, indicating that these patients need aggressive anticoagulation, and many of them required mechanical ventilation, had acute renal failure requiring dialysis and some had thrombotic stroke. Other important abnormal laboratory investigations include elevated fibrinogen with normal platelet count, elevated prothrombin time and partial thromboplastin time. Many of these patients may also present cardiac abnormalities in the form of right heart failure, right ventricle dilatation, and pulmonary thromboembolism, even after recovery from COVID-19. Some of these patients may continue to have palpitations and chest pain and MRI shows diffuse myocardial edema and fibrosis by late-gadolinium enhancement (LGE), or both along with impaired RV cardiac index and ejection fraction, despite preserved LV function. How long these abnormalities persist and whether they have any life-long consequences remain to be determined.
Neurological manifestations
In a retrospective study performed in 214 COVID-19 hospitalized patients in Wuhan, China it was reported that ~37% showed some central neurological manifestations. Some of these included headache, dizziness, impaired consciousness, ataxia, epilepsy, and stroke. Of all, the most common manifestations were headache and dizziness. Other manifestations included loss of taste and smell, neuralgia, and muscle pains, Guillain-Barre syndrome, and some of these patients showed an increase in plasma creatine kinase, implying pathological involvement of muscles. Some patients even had stroke. It is not clear how many of these neurological manifestations are truly due to nervous system involvement or because of hypoxia. Nevertheless, they need to be considered as a part of the COVID-19 manifestations. These reported neurological manifestations reported from China have been confirmed from a study done in France. In order to document properly the possible involvement of the nervous system in patients with COVID-19, detailed clinical, neurological and electrophysiological studies of these patients and efforts need to be made to isolate SARS-CoV-2 from cerebrospinal fluid, and autopsies of the COVID-19 patients need be carried out.
COVID-19 in children
Some disturbing findings reported in children need special attention. It has been reported that children are uniquely susceptible to developing acute cardiac decompensation due to severe inflammation following SARS-CoV-2 infection, termed multisystem inflammatory syndrome in children (MIS-C) [63–66]. Treatment with immunoglobulin seems to benefit these subjects with recovery of left ventricular systolic function. MIS-C has remarkably close similarities with Kawasaki disease. The data obtained so far indicate that these children present with cardiogenic shock, left ventricular dysfunction and a severe inflammatory state. The mean ages of these patients is about 10 years, all had fever, 80% had gastrointestinal symptoms such as abdominal pain, vomiting and diarrhea and comorbidities such as higher body mass index (17%), asthma (9%), lupus (3%), and overweight. Only 17% presented with chest pain. Many of these patients had left ventricular ejection fraction less than 30%, and 80% required inotropic support with 28% treated with extracorporeal membrane oxygenation (ECMO). All had a severe inflammatory state as evidenced by elevated C-reactive protein and D-dimer, higher plasma IL-6, elevated troponin, and NT-proBNP or BNP elevation. These patients are in need of IV inotropic support, IV immunoglobulin, anticoagulation with heparin, and received IV steroids.
Current therapeutic approaches to COVID-19
Several clinical trials are underway to ascertain the efficacy of several drugs that could be useful either alone or in combination for COVID-19. Some of these include interferons, guanosine-analog RNA synthesis inhibitors, reverse transcriptase inhibitors or influenza drugs, and remdesivir, an antiviral. Some drugs are being investigated for their potential to block viral entry into the host cell (umifenovir, chloroquine or interferon), viral replication (lopinavir and ritonavir, ASC09 or darunavir/cobicistat) and viral RNA synthesis (inhibitors or influenza drugs, such as remdesivir, favipiravir, emtricitabine/tenofovir alafenamide or ribavirin).
Since SARS-CoV-2 employs ACE2 and TMPRSS2 to enter target cells, attempts are being made to develop drugs that inhibit TMPRSS2. Janus-associated kinase (JAK) inhibitors that suppress inflammation are also being tested for their efficacy against COVID-19. Clinical trials are underway to use these drugs along with mesenchymal stem cells (MSCs) for COVID-19. These efforts will take time to find out which of the strategies are likely to be successful. Several vaccines are under development for SARS-CoV-2 and are likely to take considerable time before they become available for the world’s population. Furthermore, initial studies showed that those who developed COVID-19 did develop protective antibodies but their titer seemed to decline in about 3–6 months, raising the possibility that even vaccine induced antibodies may not last long. It was also noted that some of the patients who recovered from COVID-19 had anti-interferon antibodies that may interfere with the anti-viral action of IFN (interferon), which seems to be more common in those who had severe COVID-19.
In view of these uncertainties and increasing incidence of mutations, it is important to look at other novel and out-of-the-box methods of inactivating SARS-CoV-2 by understanding the natural mechanisms by which the human body fights such infections. In the current narrative review, the potential role of bioactive lipids (BALs) in the pathobiology of COVD-19 is discussed based on their actions in the regulation of inflammation and the immune response. Thus, the current study does not need ethical approval as it did not involve any interventions or studies in experimental animals or patients.
In a study that was conducted in patients diagnosed with COVID-19 and vesicular lesions wherein histological analysis and detection of severe acute respiratory coronavirus 2 (SARS-CoV-2) in the content of the vesicles was performed in 24 patients, it was reported that a disseminated pattern was found in 18 (75%) patients, and a localized pattern was found in 6 (25%) patients. The median duration of the skin rash was 10 days.
Out of the 24 patients studied, 19 (79.2%) patients presented the skin rash after the onset of COVID-19 symptoms. Histologic examination in 2 patients was consistent with viral infection, SARS-CoV-2 was not detected in 4 patients. This monocentric study shows the clinical characteristics of vesicular skin rashes in COVID-19 patients.
Bioactive lipids inactivate enveloped viruses
One of the unappreciated mechanisms by which many microbes can be inactivated by the human innate immune system is by elaboration of polyunsaturated fatty acids (PUFAs) and their metabolites (termed bioactive lipids). Several bioactive lipids, especially linoleic acid (LA), γ-linolenic acid (GLA), dihomo-GLA (DGLA), arachidonic acid (AA), α-linolenic acid (ALA), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), can inactivate streptococci, staphylococci, both Gram-positive and Gram-negative bacteria, fungi and enveloped viruses, including influenza, HCV, HBV and HIV [67–75]. These fatty acids disrupt the microbial cell membranes and interfere with the cellular metabolic processes including inhibition of respiratory activity, transportation of amino acids, and uncoupling of oxidative phosphorylation. Based on this evidence [67–75], it is reasonable to propose that alveolar macrophages, leukocytes, T and B cells, NK cells and other immunocytes release AA and other unsaturated fatty acids to inactive various microbes including SARS-CoV-2, SARS, and MERS. It is possible that the human body protects itself from various microbes using these unsaturated fatty acids. Based on these observations, it can be suggested that a deficiency of AA and other unsaturated fatty acids can predispose an individual to various infections including SARS-CoV-2, SARS, and MERS. Alveolar macrophages can secrete AA and other unsaturated fatty acids into the alveolar fluid to inactivate SARS-CoV-2, SARS and MERS and several other bacteria and fungi [76–92]. This implies that administration of these fatty acids could be of significant benefit against SARS-CoV-2, SARS, and MERS.
Phospholipase A2 (PLA2) is microbicidal in nature
Type-IIA secreted phospholipase A2 (sPLA(2)-IIA) releases AA and other unsaturated fatty acids from the cell membrane phospholipid pool, a mechanism developed by various cells including immunocytes to fight against microbes. It is noteworthy that sPLA2-IIA present in animal and human biological fluids is sufficient to bring about its antimicrobial action. Human recombinant sPLA2-IIA induced release of PUFAs in response to several bacteria and viruses at concentrations ~1.1 ng/ml that prefers anionic phospholipids such as phosphatidylglycerol, which is the main phospholipid component of bacterial membranes. In contrast to this, higher concentrations of sPLA2-IIA are needed to act on the host cell membranes and surfactant, both of which are mainly composed of phosphatidylcholine, which is a poor substrate for sPLA2-IIA. It is noteworthy that over-expression of human sPLA2-IIA renders cells resistant to Staphylococcus aureus, Escherichia coli, and Bacillus anthracis infections. Furthermore, intranasal administration of sPLA2-IIA protects mice from microbial infections including pulmonary anthrax, implying that this mode of administration of sPLA2-IIA and PUFAs could be employed to protect against SARS-CoV-2, SARS, and MERS [93–96]. In a similar fashion, even bioactive lipids can be administered intranasally to prevent various microbial infections including SARS-CoV-2, SARS, and MERS.
Bioactive lipids (BALs) including AA mediate the antimicrobial action of macrophages
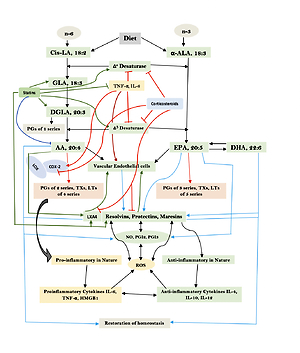
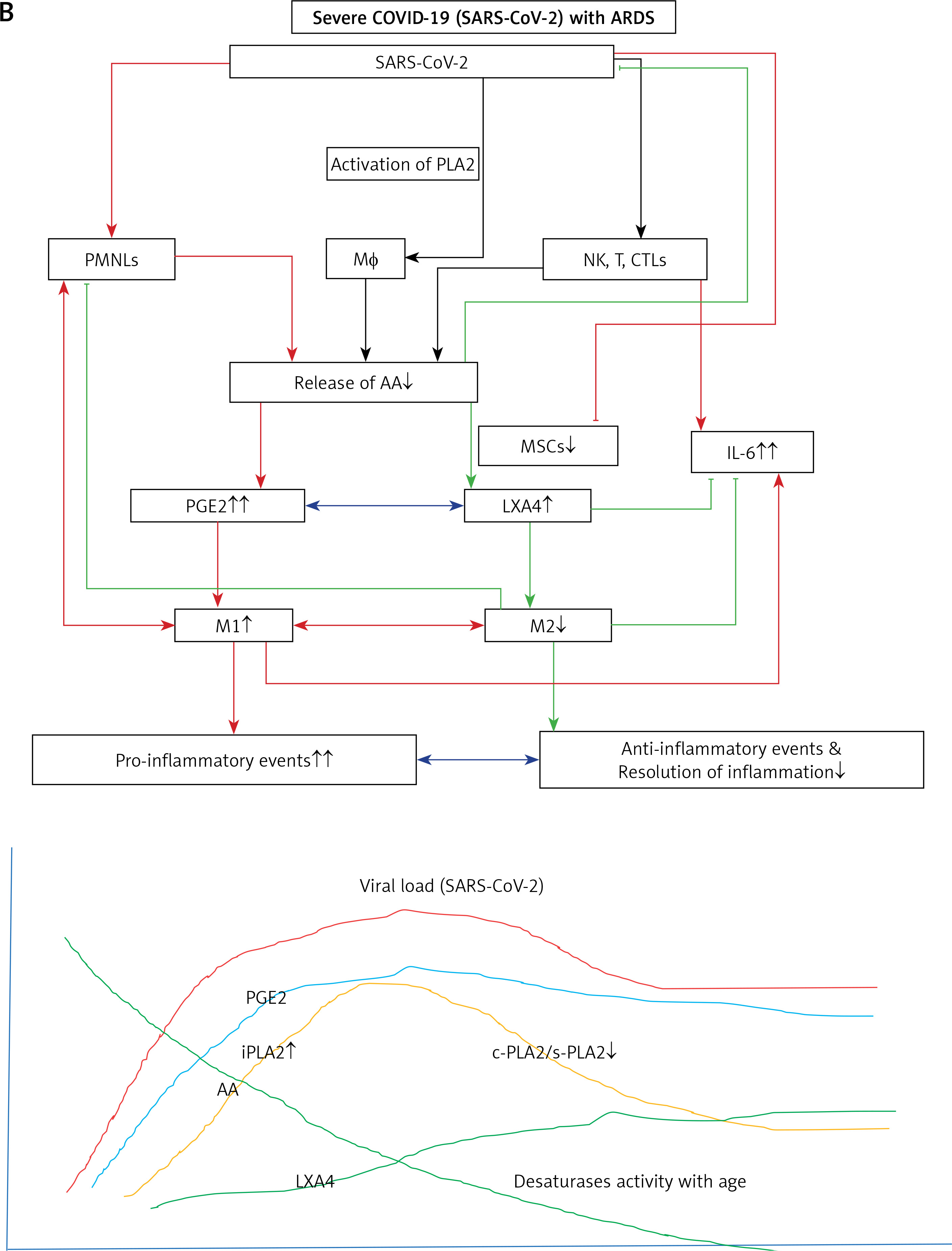
Humans are constantly exposed to various microbes through inhaled air, which calls for an efficient and simple mechanism(s) to prevent and protect lungs from various infections. In other words, alveolar macrophages need to possess an efficient mechanism to prevent microbial infections. Inhaled staphylococci can be eliminated by alveolar macrophages utilizing AA and other bioactive lipids derived from the extracellular fluid and thus protect lungs from the invading microbes. Even immunocytes may protect various tissues from microbial infections in the same fashion [72, 93, 94, 97–108] (see Figures 3 A and B for metabolism of essential fatty acids and their role in inflammation and restoration of homeostasis and their role in COVID-19). This is supported by the observation that lymphokine-activated macrophages release bioactive lipids to induce apoptosis of tumor cells [107]. Thus, alveolar lining material interacts with alveolar macrophages, whereas tumor cells interact with the microenvironment, which is tuned to the benefit of the body. Proliferation of tumor cells can be inhibited by the immunocytes present in the tumor microenvironment by secreting bioactive lipids. Even normal cells surrounding the tumor cells can secrete bioactive lipids in order to restrain tumor cell growth. At the same time, it is possible that tumor cells may take advantage of the secreted bioactive lipids, especially GLA, DGLA, AA, EPA and DHA, to form PGE2/PGE3 and leukotrienes that possess immunosuppressive actions. Thus, there may be intense competition between normal cells/immunocytes and tumor cells for the secreted bioactive lipids. The outcome (tumor cell growth or tumor cell apoptosis) of this competition depends on the way the released GLA/DGLA/AA/EPA/DHA (by the normal cells) are metabolized by normal and tumor cells. If the normal cells are able to dominate and produce lipoxins, resolvins, protectins and maresins by utilizing these fatty acids (GLA/DGLA/AA/EPA/DHA) and enhance NO and ROS generation then the tumor cells are induced to undergo apoptosis or remain dormant for a long time. In contrast, if tumor cells are able to take up these fatty acids to convert them to PGE2/PGE3/LTs and inhibit or decrease the formation/production of lipoxins, resolvins, protectins and maresins by normal cells (since the surrounding immunocytes or normal cells are unable to have access to AA/EPA/DHA), then the tumor cells will be able to induce immunosuppression and proliferate (Figure 4).
Figure 3
A – Scheme showing the metabolism of essential fatty acids, their role in inflammation and cytoprotection of endothelial cells. Note that corticosteroids block the activity of PLA2, desaturases, COX-2 and LOX and thus cause EFA/PUFA deficiency and inhibit the formation of PGE2, LTs and of LXA4/resolvins, protectins and maresins. Statins enhance the activities of desaturases and increase the formation of GLA, DGLA, AA and of LXA4/resolvins/protectin/maresins. TNF-α and IL-6 decrease the activity of desaturases and thus decrease the formation of GLA, DGLA and AA but enhance the activity of COX-2 and thus cause EFA/PUFA deficiency but increase the formation of PGE2. TNF-α and IL-6 stimulate PLA2 and thus increase the release of AA from the cell membrane lipid pool. Thus, both corticosteroids and TNF-α and IL-6 cause an EFA/PUFA deficiency state but have opposite actions on PLA2 and PGE2 formation B – Scheme showing the relationship among SARS-CoV-2 and other enveloped virus induced events on EFA metabolism, pro- and anti-inflammatory cytokines and their modulation by steroids and statins. For details see text. Modified from Das UN. J Inflammation Res 3: 143-70
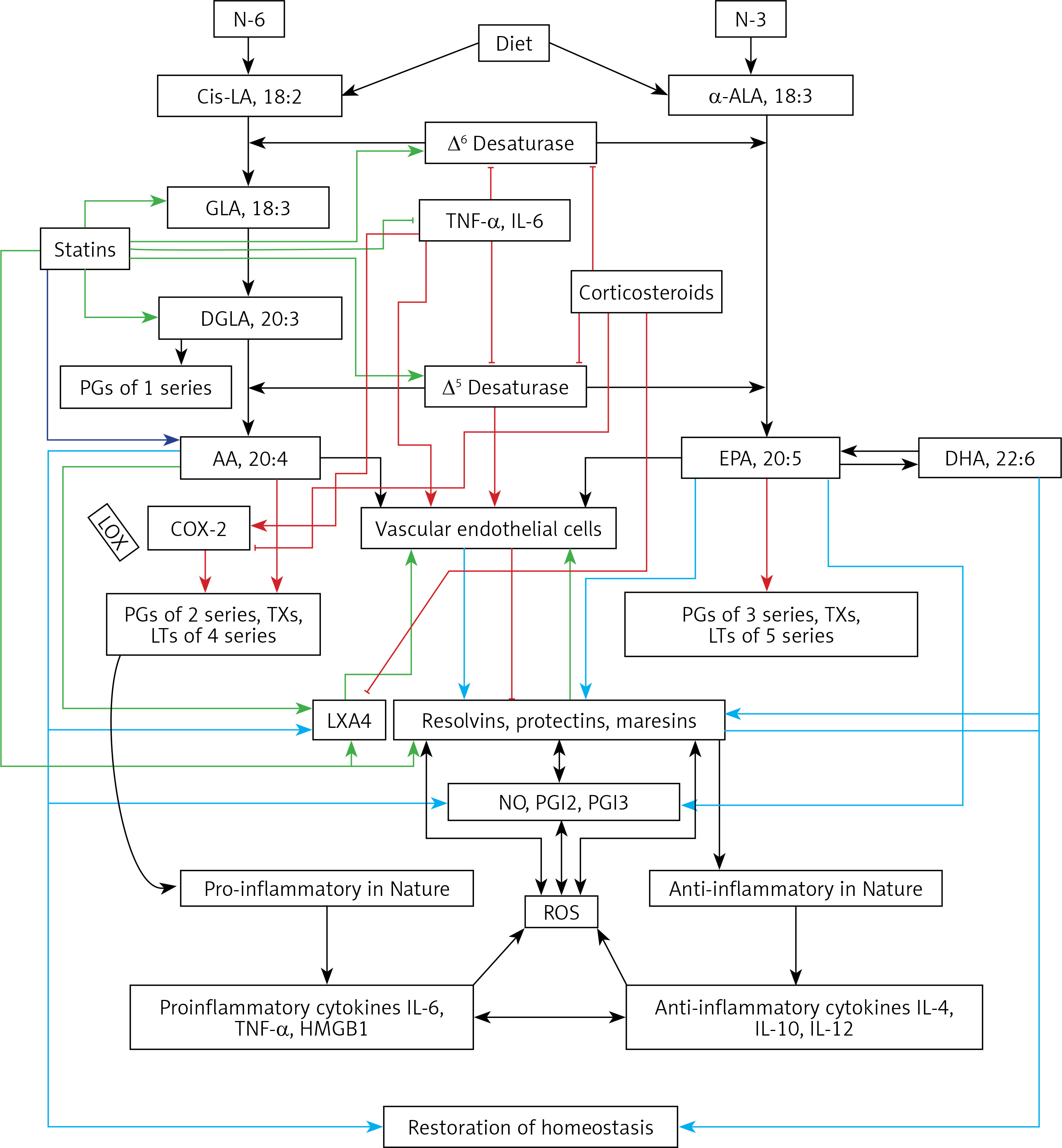
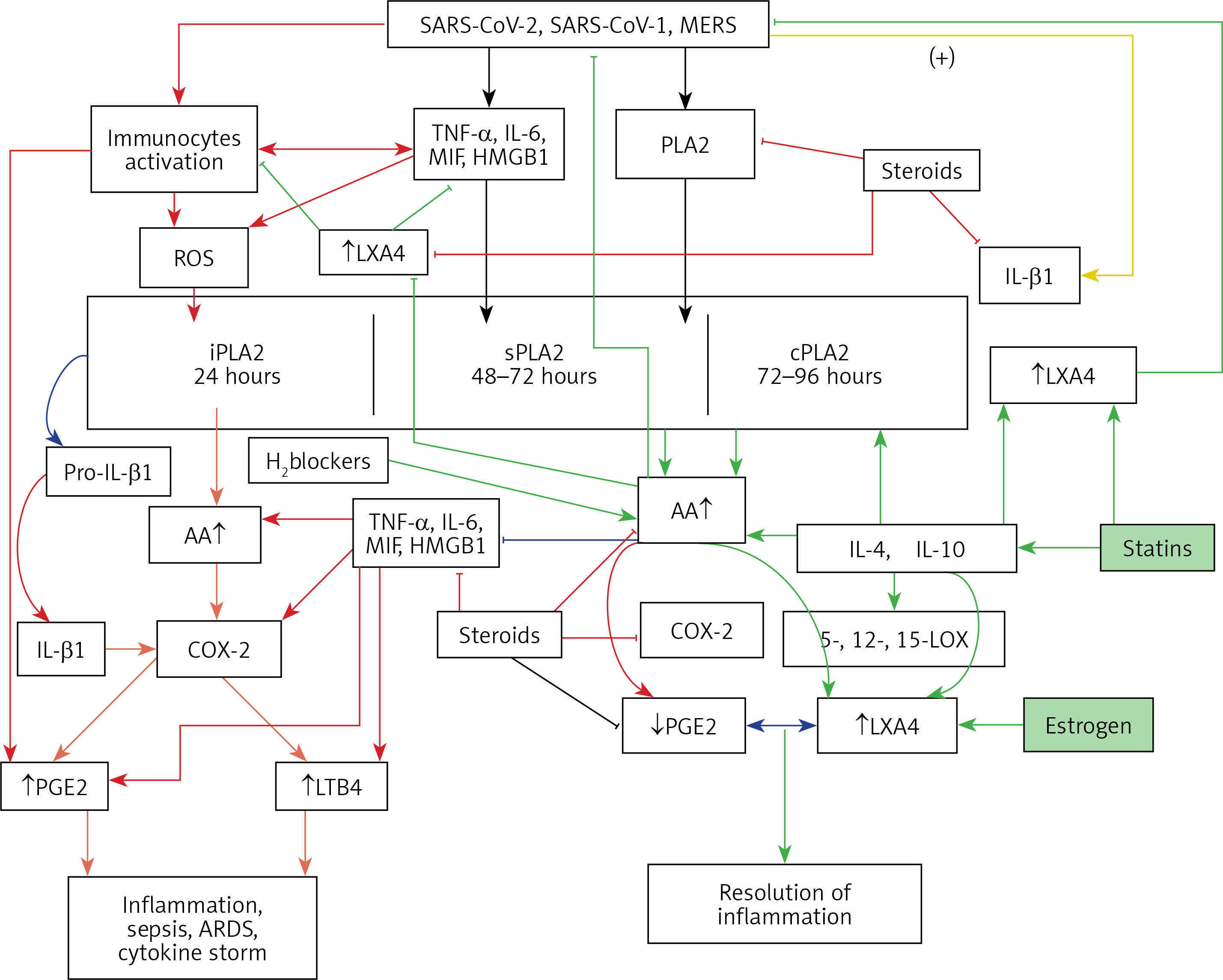
Figure 4
A – Scheme showing potential interaction(s) among invading microbes including SARS-CoV-2, host cell (target tissue/cell), immunocytes including macrophages and bioactive lipids including GLA/DGLA/AA/EPA/DHA, LXA4, PGE2, lipid peroxides (LP), activity of PLA2 COX-2 and LOX enzymes, CO, NO, H2S, and soluble epoxide hydrolase (sEH) and their relationship to the ability of microbes to infect the host cell and the response of the host cell to infection. B – Similarly, potential interaction between tumor host cell (refers to surrounding normal cells present in the microenvironment of tumor) and the role of COX-2, LOX enzymes and the formation of PGE2/LXA4 and their effect(s) on tumor cell. Legend to Figure 4: A – When a microbe (including SARS-CoV-2) invades a normal cell (host cell), it may increase the production of PGE2 using its own (or host cell) COX-2. The microbe using its LPS or by activating host cell LOX enzymes leads to the formation of LXA4 that negates the synthesis or action of PGE2. Microbial infection activates immunocytes leading to release of IL-6 and TNF-α that enhance the production of NO, CO, H2S and other ROS that can kill the microbes. Microbes can also activate PLA2 that induces the release from the cell membrane lipid pool of AA/EPA/DHA that are utilized for the formation of PGE2, LXA4 and lipid peroxides. Depending on the stage of the inflammation initially there will be activation of iPLA2 (inducible phospholipase A2) that induces the release of AA that is directed to form PGE2 and activation of M1 resulting in inflammation. Once PGE2 levels reach a peak it triggers the activation of soluble PLA2 (sPLA2) and cytosolic PLA2 (cPLA2) inducing the release of a second wave of AA and simultaneously activation of LOX leading to the formation of LXA4 and M2. This results in suppression of inflammation and restoration of homeostasis. BALS especially AA/EPA/DHA/LXA4 and lipid peroxides kill the microbes including SARS-CoA2. Lipid peroxides, LXA4 and AA/EPA/DHA may inhibit the activity of soluble epoxide hydrolase that leads to suppression of inflammation. Mesenchymal stem cells (MSCs) produce LXA4 to bring about their beneficial actions. B – A similar set of events occurs in the presence of a tumor cell. There is a crosstalk between normal cells surrounding the tumor cell and immunocytes. Tumor cells have high COX-2 activity that may lead to increased formation of PGE2 (COX-2 of tumor cells may act on normal cells or on exposure to tumor cells normal cells may enhance their COX-2 activity). On exposure to the tumor cell, PLA2 of normal cells/immunocytes is activated to induce the release of AA/EPA/DHA to produce PGE2 or LXA4. If the released AA/EPA/DHA are converted to form excess lipid peroxides and/or LXA4, then tumor cell growth is inhibited or it undergoes apoptosis. If the activity of COX-2 dominates then AA is converted to PGE2 that induces immunosuppression and enhances tumor cell growth. IL-6 and TNF-α released by immunocytes induce the release of ROS that can enhance the formation of lipid peroxides to induce apoptosis of tumor cells. But both IL-6 and TNF-α can induce deficiency of AA/EPA/DHA in the tumor cells and surrounding normal cells and immunocytes by inhibiting the activities of desaturases. See text for further details.
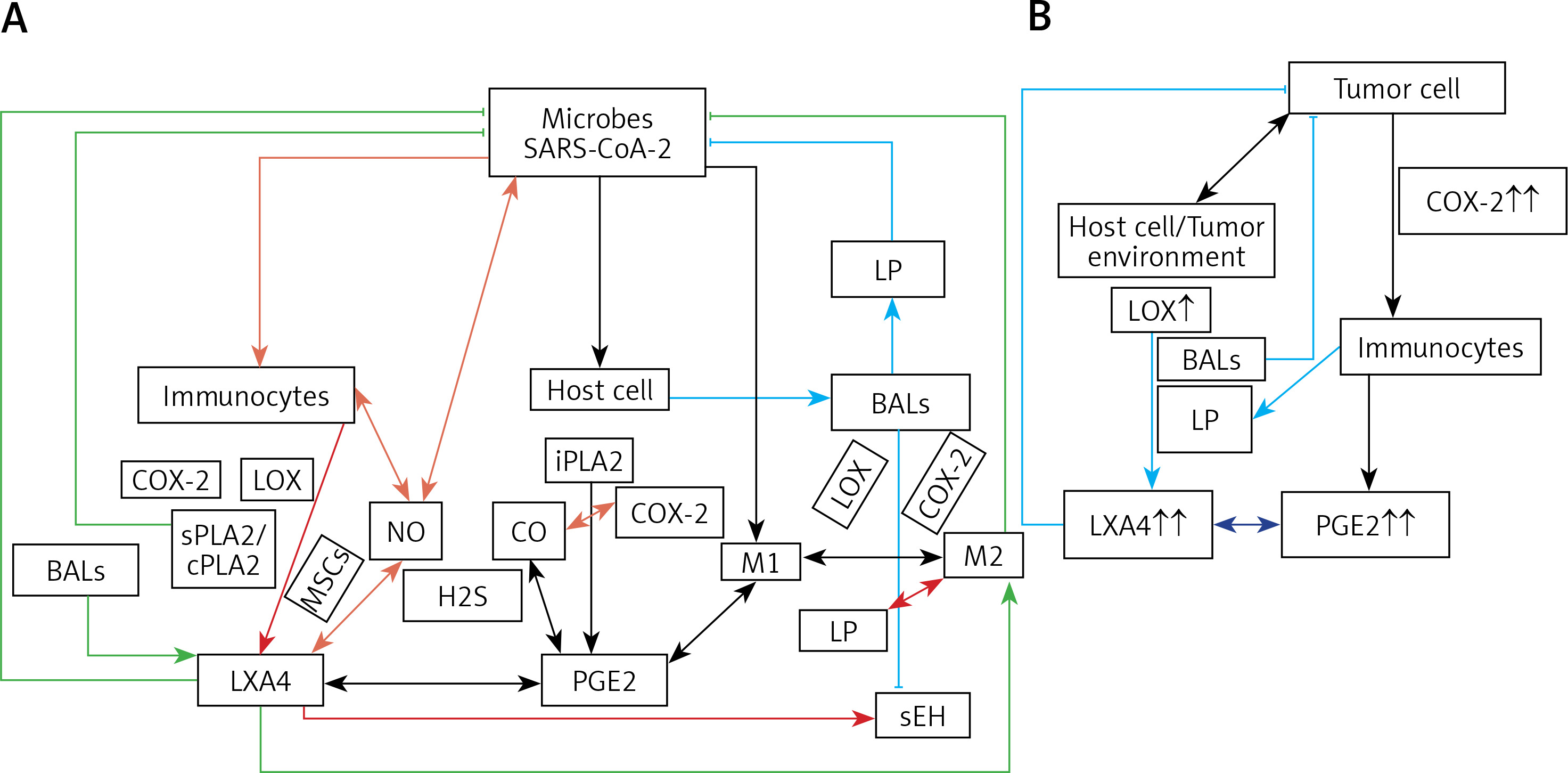
Crosstalk between microbes and host cells/tissues
Similarly, there could occur a crosstalk between the invading microorganisms and the host tissue/cells to utilize GLA/DGLA/AA/EPA/DHA secreted by normal cells/target cells (including T cells and NK cells) and the microbes. Microbes are known to influence the metabolism of GLA/DGLA/AA/EPA/DHA by target cells to form PGE2/PGE3/LTs to produce local inflammation and immunosuppression. On the other hand, the host tissues/cells and immunocytes utilize these fatty acids to inactivate microbes by producing lipid peroxides, NO, and regulating the production of IL-6, TNF-α and IFN and other anti-microbial cytokines/products. This is supported by the observation that (i) some bacteria can induce activation of the inflammatory signaling pathway [109]; (ii) COX-2 expression was increased in macrophages infected with the wild-type colon cancer-associated E. coli 11G5 strain but not by a clbQ mutant, indicating that tumor-infiltrating bacteria induce the expression of COX-2 to enable the tumor cells and tumor infiltrating macrophages to produce PGE2, a immunosuppressor [110]; (iii) heat-killed Mycobacterium can downregulate MMP-9 in murine peritoneal macrophages through COX-2 and independent of TNF-α [111], suggesting that immunosuppression seen in the host during tuberculosis infection is due to local production of PGE2; and finally, (iv) COX-2 can be induced by cytomegalovirus (CMV) in human RPE cells, which can lead to translocation of the induced NF-κB from the cytoplasm to the nucleus associated with an increase in the production of PGE1 and PGE2 in these cells that significantly increased CMV proliferation [112]. Since, under physiological conditions, there is a balance maintained between pro-inflammatory PGE2 and anti-inflammatory lipoxins, resolvins, protectins and maresins, it is reasonable to suggest that these anti-inflammatory bioactive lipids inhibit viral replication (similar to GLA/DGLA/AA/EPA/DHA). These studies lend support to the concept that microbes (and in a similar fashion tumor cells) enhance the expression of COX-2 and other enzymes of the PG pathway in the host cell and thus alter the balance between pro-inflammatory and anti-inflammatory bioactive lipids at the site of infection or tumor invasion. This, in turn, alters the host response to tumor cells or infection by the invading microbes (including viruses such as SARS-CoV-2). It is possible, but yet to be verified, that tumor cells can upsurge the COX-2-LOX pathway of the normal cells in the tumor microenvironment for their benefit. In a similar fashion, certain bacteria (such as Lactobacillus) may support the host tissues to overcome microbial resistance by modulating host COX-LOX enzymes [113, 114].
Immunocytes release bioactive lipids (BALs)
Immunocytes (NK cells, cytotoxic tumor lymphocytes (CTLs)), lymphokine activated killer cells, dendritic cells, leukocytes, etc.) release perforin and granzyme, and the cytokines IL-6, TNF-α, and IFN-γ to eliminate tumor and infected cells. Surprisingly, NK cells and CTLs induce tumor cell apoptosis even in the absence of perforin and granzyme by enhancing the expression of soluble PLA2 (sPLA2) [115]. PLD (phospholipase D) activation that leads to release of AA is needed for the CD16-triggered signaling cascade to induce exocytosis of the NK cytotoxic granules [116, 117]. It has been reported that lipids form a constitutive component of cytolytic granules of CTL, NK and γδT cells [118] and cytokine-activated macrophages (and probably other immunocytes) can release bioactive lipids to induce apoptosis of tumor cells [107, 119–121]. These results emphasize the importance of PLA2/PLD and release of bioactive lipids for the cytolytic action and release of cytolytic granules by immunocytes.
Macrophages and tissue-resident memory CD8+ T cells cooperate with each other to sense pathogens and protect the barrier tissues [122]. This is somewhat akin to the crosstalk/interaction seen between alveolar lining material and macrophages to eliminate invading microbes and tumor cells [100]. A close cooperation/interaction exists between epithelial cells and macrophages that is similar to the interaction between tissue resident memory T cells and their surrounding milieu. The resident memory T cells are dependent on fatty acids for their survival and function. These much-needed fatty acids are transported to the T cells by the specific fatty acid binding protein (FABP). The type of FABP expressed by the T cells depends on the tissue in which they are resident, implying that the tissue resident memory T cells show varying patterns of FABP isoform usage that, in turn, is determined by tissue-derived factors. To meet the fatty acid demands of the tissue resident memory T cells (and other immunocytes including macrophages), these cells modify their FABP expression depending on the tissue in which they are located or relocated [123]. This suggests that each tissue and its resident memory T cells (and macrophages and other immunocytes) need to get a specific type(s) of bioactive lipid(s) not only for their survival but also to bring about their specific action(s) that is tailored to their location as dictated by the local milieu. In an extension of this argument, it can be postulated that to inactivate a specific type of invading microbes (including viruses) a specific type or types of bioactive lipid(s) is needed that, in turn, depends on the target tissue.
Macrophages can be either M1 or M2 type. M1 type macrophages have pro-inflammatory actions whereas M2 is anti-inflammatory in nature (Figure 5). M1 macrophages are needed to kill the invading microbes including SARS-CoV2 and clear the debris. Once the much-needed inflammatory process is completed and invading microbes are cleared, M2 macrophages are needed to induce resolution of inflammation and restore homeostasis. PGE2 and leukotrienes are needed for the generation of M1 macrophages and they (M1) generate these pro-inflammatory PGE2 and leukotrienes. In contrast, anti-inflammatory cytokines and lipoxin A4 (LXA4) from AA, resolvins from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) and protectins and maresins from DHA facilitate generation of M2 macrophages and induce resolution of inflammation, restoring homeostasis [73, 124–136]. But there is one caveat for this process of inflammation to anti-inflammation or resolution of inflammation to occur in a coordinated, smooth and orderly fashion, which is the availability of GLA/DGLA/AA/EPA/DHA in adequate amounts at the right time and at the right place. Any discrepancy in this sequence of events may result in the acute inflammatory process becoming chronic and failure of resolution. This smooth transition from pro-inflammatory events to resolution of inflammation and restoration of homeostasis needs adequate activity of desaturases, COX and LOX enzymes (including soluble epoxy hydrolase: sEH), PGDH, an appropriate balance between pro- and anti-inflammatory cytokines, generation of adequate amounts of ROS, NO, CO, H2S and sufficient activity of various anti-oxidants. These enzymes, free radicals and antioxidants are needed to regulate oxidative and mitochondrial stress and to regulate the actions of leukocytes, macrophages, T cells, NK cells and CT cells and to process smooth and timely communication(s) between various immunocytes, host cells/tissues and the invading microbes (including SARS-CoV-2). Any hindrance of any one of these processes will result in exacerbation of the clinical picture of COVID-19 and lead to the onset of its complications.
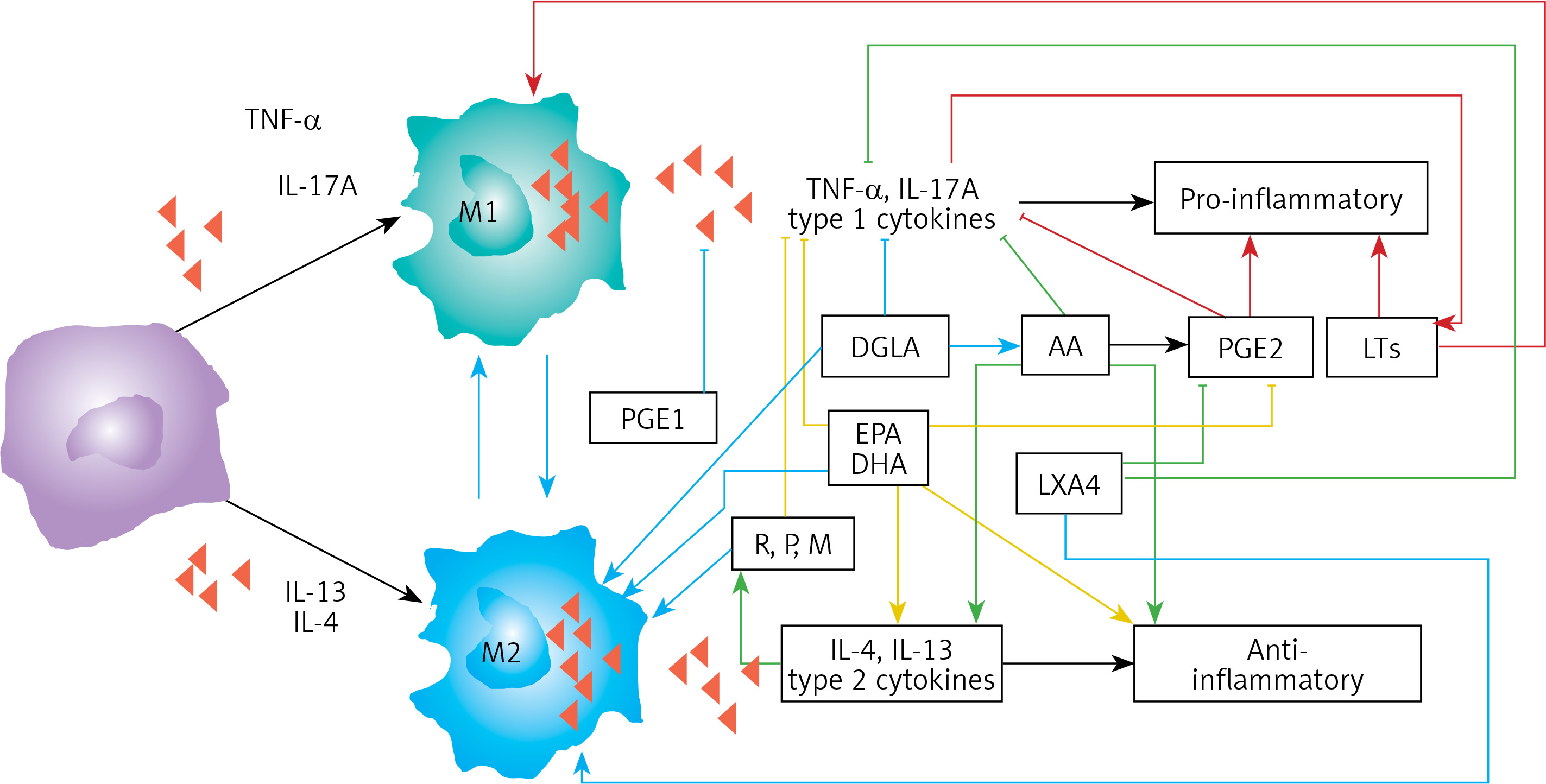
Figure 5
Scheme showing M1 and M2 macrophages and various cytokines produced by them and their respective functions. DGLA, AA, EPA and DHA have anti-inflammatory actions and inhibit the production of pro-inflammatory TNF-α, IL-2 and IL-1 and facilitate the generation of M2 macrophages. PGE1 formed from DGLA, LXA4 from LXA4 are anti-inflammatory in nature. Resolvins (R), protectins (P), and maresins (M) formed from EPA and DHA are anti-inflammatory and block the production of TNF, IL-1, IL-2. PGE2, leukotrienes B4, D4 and E4 formed from AA are pro-inflammatory in nature. LXA4, resolvins, protectins and maresins inhibit the production of PGE2 and LTs. Leukotrienes (of 5 series) are also formed from EPA that have pro-inflammatory action but are much less potent compared to LTs formed from AA. EPA and DHA inhibit the production of PGE2
In this context, the relationship between pro-inflammatory cytokines and EFA metabolism needs a closer look. Both TNF-α and IL-6 can block the activities of desaturases, which are needed for the conversion of dietary linoleic acid (LA) and α-linolenic acid (ALA) to their respective long-chain metabolites, namely AA and EPA and DHA respectively [137]. This could be the reason why patients with rheumatoid arthritis, lupus, and sepsis have low plasma concentrations of AA, EPA and DHA [138–141]. As a consequence of this precursor (GLA, DGLA, AA, EPA and DHA) deficiency, the generation of LXA4, resolvins, protectins and maresins that are potent inhibitors of IL-6 and TNF-α formation is insufficient. Hence it is predicted that patients with COVID-19 will have deficiency of GLA/DGLA/AA/EPA/DHA, LXA4/maresins, and resolvins/protectins. These results indicate that administration of GLA/DGLA/AA/EPA/DHA/PGE1/LXA4, resolvins, protectins and maresins will be of significant benefit in COVID-19 by (i) suppressing inappropriate production of IL-6 and TNF-α, (ii) ameliorating inflammation and excess pro-inflammatory cytokine storm, and (iii) protecting normal cells (especially bone marrow cells, myocardial cells, hepatic and renal tissues) and thus initiate recovery [142–155].
Bioactive lipids and M1 and M2 macrophages
Once the inflammatory process has achieved its function of eliminating the invading microbes (including SARS0CoV-2) and debris removal is completed by the M1 type, macrophages change their phenotype to M2 type to initiate resolution of the inflammatory process by producing anti-inflammatory cytokines and anti-inflammatory bioactive lipids such as lipoxin A4 (LXA4) from AA, resolvins from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) and protectins and maresins from DHA [156–169]. These results imply that pro-inflammatory bioactive lipids induce the M1 phenotype whereas anti-inflammatory bioactive lipids trigger the generation of M2 macrophage phenotype. These studies suggest that the way GLA/DGLA/AA/EPA/DHA are metabolized by the macrophages and their modulation by the pro- and anti-inflammatory cytokines and changes in the activities of desaturases, cyclo-oxygenase-2 (COX-2) and LOX (5-, 12-, and 15-lipoxygenases) determines the shift in the balance between M1 and M2 macrophages [156–169]. Thus, generation of adequate concentrations of GLA/DGLA/AA/EPA/DHA and their conversion to PGE1, LXA4, resolvins, protectins and maresins by the appropriate concentrations and action of desaturases, COX-2, 5-, 12-, and 15-LOX and 15-PGDH result in the generation of M1 and M2 macrophages as the situation demands. When the adequate amounts of IL-6 and TNF-α and other pro-inflammatory cytokines are generated in the initial stages of infection due to SARS-CoV-2 (and other enveloped viruses and microbes) and formation of PGE2 occurs in adequate amounts it leads to the generation of M1 macrophages, whereas generation of M2 type occurs when PGE1/LXA4/resolvins/protectins/maresins are formed in appropriate amounts at the time of resolution of inflammation [156–169]. Though this argument seems simple and straightforward, it is not known how exactly this shift of balance from the pro- to the anti-inflammatory pathway and M1 to M2 phenotype occurs. In this shift of balance from the pro- to the anti-inflammatory pathway and maintenance of balance between M1 and M2 macrophages, there seems to be a critical role for NO/CO/H2S (nitric oxide/carbon monoxide/hydrogen sulfide), which have an important role in the acceleration of resolution of inflammation and tissue regeneration. This balance between pro- and anti-inflammatory status and M1 and M2 macrophages and factors that could be involved in their formation and action are critical for fighting viral infections HCV, HBV, SARS-CoV-2, SARS and MERS and restoring homeostasis by inducing necessary tissue repair and regeneration and resolution of inflammation (Figures 3 B, 4, 5, and 6 A).
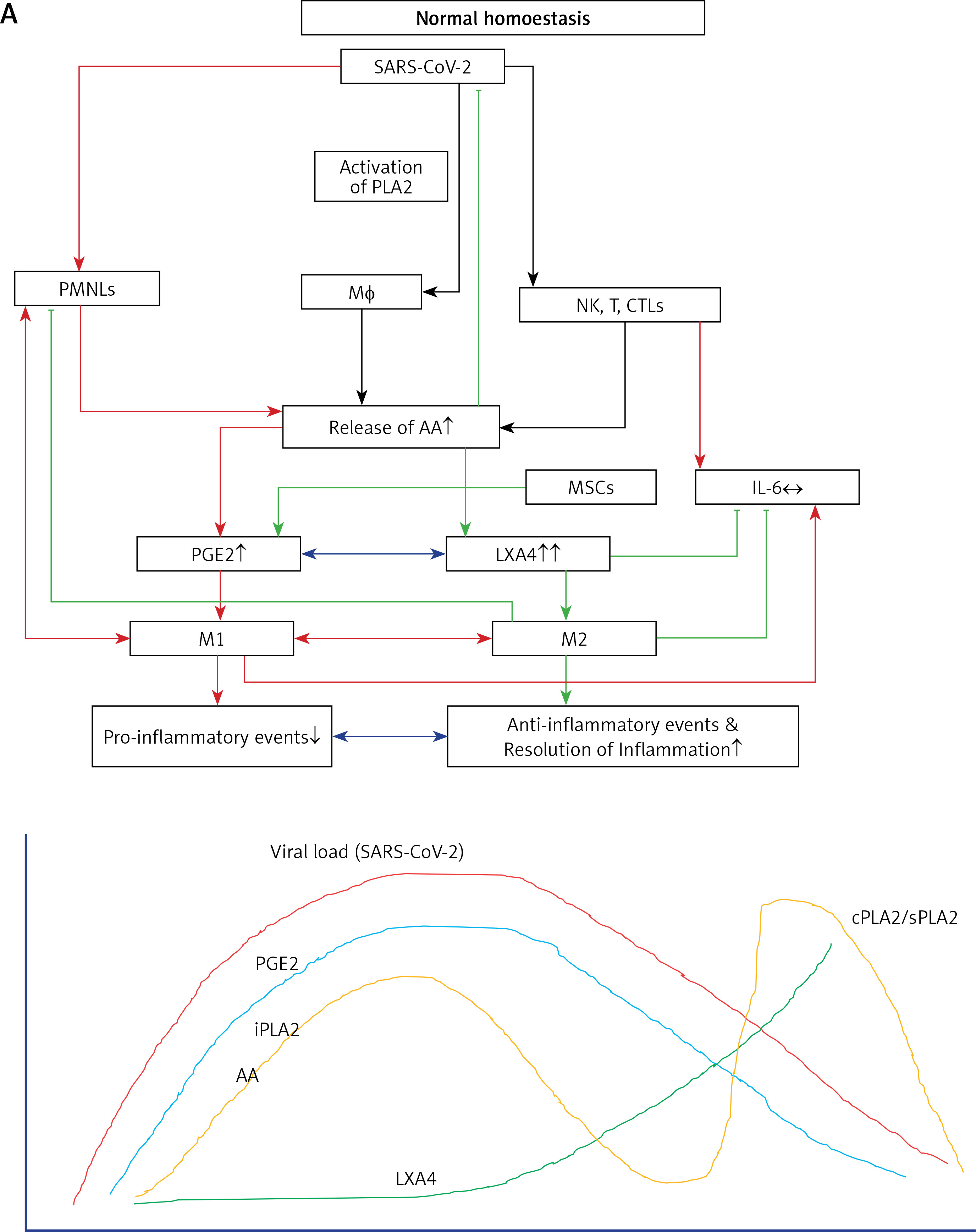
Figure 6
A – Scheme showing potential relationship among AA, PGE2, LXA4 and viral load in a COVID-19 patient who recovers. AA is released from the cell membrane in two phases; the first phase is used for PGE2 synthesis, whereas the second phase is meant for LXA4 synthesis. Once PGE2 concentration reaches a peak, LXA4 synthesis is triggered, which induces resolution of inflammation. AA release is triggered by SARS-CoV-2 and other infections B – Scheme showing potential relationship among AA, PGE2, LXA4 and viral load in a COVID-19 patient who succumbs to the disease. Absence of biphasic nature of AA release and failure of PGE2 to reach a peak to trigger LXA4 synthesis that results in failure of resolution of inflammation (compare with Figure 7 A). DGLA, EPA and DHA (not shown in the figure) may have actions/functions like AA. Desaturases activities fall with age
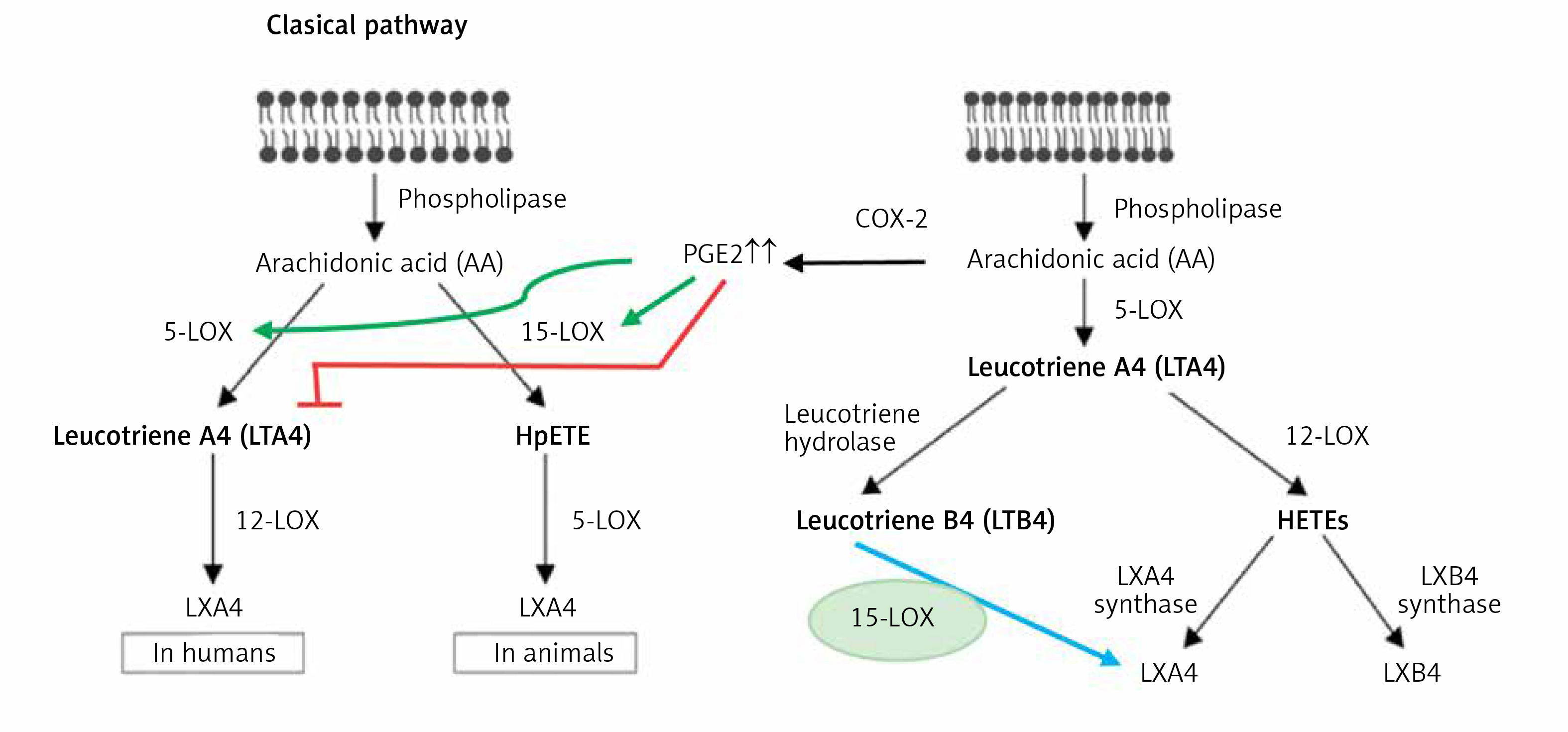
Interaction between PGE2 and LXA4
Despite the fact that, in general, PGE2 is considered as a pro-inflammatory bioactive lipid, studies did suggest that this prostaglandin can also have anti-inflammatory and immunosuppressive actions [170–176], especially by altering macrophage polarization by MSCs [177–179]. These pro- and anti-inflammatory actions of PGE2 seem to depend on its binding to different types of PGE2 receptors (EPs) that is concentration dependent. At low concentrations, PGE2 binds to the high-affinity EP4 receptor and enhances IL-23 production [180], whereas large amounts of PGE2 bind to the EP2 receptor and suppress IL-23 release [181, 182]. These results account for the dual pro- and anti-inflammatory actions of PGE2 [183]. Furthermore, PGE2 can induce LXA4 and suppress LTB4 production by modulating 5- and 15-lipoxygenase expression and thus enable the pro-inflammatory status to be switched over to the anti-inflammatory pathway (see Figure 7). This switchover from the pro-inflammatory (LTB4) pathway to the anti-inflammatory pathway (LXA4) enables the removal of neutrophils from the site of inflammation and helps in the resolution of inflammation [182–184]. This crosstalk between macrophages and neutrophils on one hand and PGE2 and LXA4 on the other enables induction of resolution of inflammation that is critical to dampening inflammation. This redirection of the formation of PGE2 from AA to LXA4 to induce resolution of inflammation seems to depend on the biphasic release of AA from the cell membrane lipid pool. The first pulse of release of AA occurs due to the activation of iPLA2 (calcium independent phospholipase A2) that is utilized for the formation of PGE2, whereas the second phase of release of AA due to the activation of cPLA2 (cytosolic phospholipase A2) and sPLA2 (secreted phospholipase A2) is utilized for the formation of LXA4 (Figures 5–7). This implies that the cell has the unique ability to direct AA released by iPLA2 to form PGE2 and cPLA2 and sPLA2 induced release of AA for the formation of LXA4. It is not clear whether other PGEs also have actions similar to PGE2. For instance, PGE1, derived from DGLA, is an anti-inflammatory, vasodilator, and platelet anti-aggregator, whose actions are remarkably similar to those of LXA4. It is likely that PGE1 may also enhance the formation of LXA4. But this needs to be confirmed.
PGE2 augments tissue regeneration
It is evident from these results [161–184] that once the inflammation and the concentrations of PGE2 reach a peak, AA released from the cell membrane lipid pool is redirected to form LXA4 instead of PGE2 (Figures 3 B, 4 and 7). This redirection of AA metabolism from PGE2 to LXA4 is an essential step needed to induce resolution of inflammation [156, 183–185]. This attainment of peak PGE2 concentration not only initiates LXA4 synthesis but also simultaneously triggers tissue regeneration. This is supported by the report that 15-PGDH (15-prostaglandin dehydrogenase, a prostaglandin degrading enzyme)-deficient mice not only have a twofold increase in bone marrow, colon, and liver PGE2 levels but also showed increased fitness of these tissues with augmented hematopoietic capacity. These 15-PGDH deficient animals showed rapid liver regeneration after partial hepatectomy and enhanced recovery of neutrophils, platelets, and erythrocytes [185]. Several other studies are in support of this observation that PGE2 promotes hematopoiesis [186–189]. Thus, administration of PGE2, LXA4, AA, EPA and DHA, resolvins, protectins and maresins, which are potent inhibitors of IL-6, TNF-α and other pro-inflammatory cytokines [154–156, 182, 183], is expected to be of significant benefit in COVID-19. It is predicted that administration of appropriate amounts and in a timely manner of AA/PGE2/LXA4 and other bioactive lipids not only will inactivate SARS-CoV-2 virus [117, 133, 135, 156, 163, 190, 191] but can prevent or even reverse inappropriate activation of coagulation, microvascular thrombosis and consumption of coagulation factors seen in those with severe COVID-19 and suppress the cytokine storm [183, 191].
LXA4 mediates MSC resolution of inflammation
It has been demonstrated that bone marrow derived MSCs can reduce the severity of acute lung injury by secreting LXA4, whereas blocking the LXA4 receptor reversed this protective action. Furthermore, LXA4 by itself has been shown to suppress LPS-induced acute lung injury in experimental animals by decreasing the production of TNF-α and MIP-2. Both human alveolar epithelial type II cells and MSCs express biosynthetic enzymes and receptors for LXA4 [192, 193]. These results coupled with the observation that MSCs are effective in the treatment of diabetic nephropathy by secreting LXA4 that, in turn, downregulates TNF-α, IL-6, IL-8, and IFN-γ [194] may explain the beneficial action of MSCs reported in COVID-19 [195].
In addition, targeted PGE2 suppression has been shown to significantly improve survival against lethal influenza infection, whereas PGE2 administration enhanced mortality [196]. These results can be interpreted to indicate that when PGE2 synthesis is blocked in a specific fashion, there will be an increase in LXA4 synthesis since both PGE2 and LXA4 are derived from the same precursor AA that, in turn, is responsible for the beneficial action of inhibition of PGE2 pathway and thus an increase in LXA4 formation is of benefit in influenza and COVID-19 (Figures 3 B, 4, 6, 7). It is noteworthy that AA can also be metabolized by the cytochrome P450 set of enzymes to form a variety of epoxyeicosatrienoic acids (EETs) that are also known to possess anti-inflammatory action. EETs are degraded by soluble epoxy hydrolase (sEH) and hence inhibition of sEH will enhance their half-life and thus their beneficial actions can be augmented. It was reported that LXA4 inhibits the activity of sEH and supplementation of ALA (and possibly other fatty acids such as AA/EPA/DHA) was found to inhibit the activity of sEH [197, 198]. These results suggest that AA/EPA/DHA/LXA4/resolvins/protectins and maresins inhibit sEH activity and ultimately enhance the concentrations of LXA4/resolvins/protectins/maresins and possibly inhibit excess production of PGE2 and thus suppress inflammatory events seen in COVID-19 (Figures 3 B, 4, 6, 7).
Increased mortality due to COVID-19 in those with co-morbid conditions and BALs
It is known that those who have obesity, type 2 diabetes mellitus, hypertension, and coronary heart disease and even otherwise healthy elderly have a risk of mortality if they get COVID-19. The exact cause of this is not clear. Previously, we found that plasma levels of AA/EPA/DHA are low in these diseases, suggesting that the activities of desaturases are defective or decreased, leading to a deficiency of AA/EPA/DHA in them [199]. In addition, these fatty acids are needed to maintain normal cell membrane fluidity and integrity. Hence, AA/EPA/DHA deficiency will lead to alterations in the expression of ACE2 and SARS-CoV-2 cannot be inactivated by BALs (since their concentrations are below the physiological optimum levels), leading to an increase in the virulence of the virus. AA/EPA/DHA deficiency induced alterations in the cell membrane fluidity may enhance the binding of SARS-CoV-2 to ACE-2. This is supported by the recent report that SARS-CoV-2 spike (S) receptor binding domains (RBDs) tightly bind the essential free fatty acid (EFA) linoleic acid (LA) in three composite binding pockets. LA binding appears to stabilize a locked S conformation that results in reduced ACE2 interaction in vitro. In human cells, LA supplementation synergized with the COVID-19 drug remdesivir and suppressed SARS-CoV-2 replication [200]. It is likely that other PUFAs such as GLA, DGLA, AA, EPA and DHA may have similar properties and thus selectively inhibit the binding of SARS-CoV-2 to ACE2 and inhibit viral replication as suggested previously [156, 183, 190, 191]. In such a scenario, deficiency of AA/EPA/DHA will also result in decreased formation of LXA4/resolvins/protectins/maresins and as a result the inflammatory process triggered by SARS-CoV-2 will flourish, leading to cytokine storm.
Several studies have suggested that children are unlikely to have severe COVID-19 and even if they develop the disease it is rather mild, unlike in the elderly, who may have the cytokine storm and consequently a higher degree of mortality. This discrepancy in the clinical picture and mortality due to COVID-19 remains unexplained but may also be attributed to BALs. It is known that with increasing age there is a decrease in the activity of desaturases. Thus, the elderly will have low activity of delta-6-desaturase and delta-5desaturase and so have a decrease in the plasma levels of GLA/DGLA/AA/EPA/DHA. In addition, with increasing age there is a fall in the plasma circulating levels of LXA4 [201]. Thus, deficiencies of AA/EPA/DHA and LXA4 and low activities of desaturases may account for severe COVID-19 seen in the elderly and those with other co-morbid conditions. This could be rectified to a large extent by supplementation of AA/EPA/DHA. This is supported by the observation that supplementation/administration of AA does not increase the formation of PGE2 and inflammation but on the contrary may suppress inflammation [202–204]. AA can be given both orally and intravenously without any significant side effects and is safe for normal healthy subjects [205].
Young children are unlikely to have significant COVID-19 since they have high activity of desaturases and are able to form significant amounts of AA/EPA/DHA/LXA4 when needed (Figure 6). The robustness of the desaturase-PLA2-BALs pathway in the children may render them less susceptible to the cytokine storm and COVID-19 infection and when this system fails, they may develop systemic multi-system inflammatory syndrome characterized by myocarditis, ARDS, and other features described in them.
Conclusions and therapeutic implications
It is evident from the preceding discussion that AA and other PUFAs can potentially inactivate SARS-CoV-2, SARS, MERS, HCV, HBV, and influenza viruses. Of all the unsaturated fatty acids, perhaps AA (AA > EPA = DHA) is the most potent in this ability. Studies suggested that AA need to undergo peroxidation to inactivate HCV and HBV since vitamin E that interferes with its (AA) peroxidation abrogated its (AA) anti-viral action [73–75, 157, 182].
Inflammatory events triggered by microbes including SARS-CoV-2 result in excess production of IL-6 and TNF-α and PGE2 and LTs by leukocytes, macrophages, T cells and the target tissues that can be suppressed by BALs GLA/DGLA/AA/EPA/DHA and their metabolites LXA4, resolvins, protectins and maresins. Thus, both inactivation of enveloped viruses and suppressing the associated inflammatory events is achieved by these BALs in association with anti-inflammatory cytokines. This implies that a delicate balance normally exists between pro- and anti-inflammatory events and molecules involved in these processes that ultimately determines recovery and restoration of homeostasis. In addition, BALs also can regulate the plasticity of macrophages to enable them to switch from M1 to M2 phenotype (or vice versa) (Figure 5). Based on this evidence, it is reasonable to propose that administration of AA/EPA/DHA could be of significant benefit in SARS-CoV-2, SARS, and MERS (and in HCV and HBV and influenza) infections. These PUFAs could be administered orally and/or intravenously without any significant side effects. It is not unreasonable to suggest that suitable preparations of PUFAs could be prepared for their intranasal administration.
Recent studies did suggest that formation of neutrophil extracellular traps (NETs) may contribute to organ damage and mortality in COVID-19 [206]. It is noteworthy that NET formation can be modulated (their formation can be enhanced or suppressed as the situation demands) by LXA4/resolvins/protectins/maresins [207–210] – yet another mechanism by which BALs are of potential therapeutic benefit in COVID-19. BALs are also capable of inhibiting Janus kinase (JAK) and thus may bring about their beneficial action in COVID-19 [211–214]. Thus, BALs have several actions that attest to the fact that they are of benefit in COVID-19. In addition, corticosteroids are known to inhibit the activities of PLA2, desaturases, COX-2 and LOX enzymes and thus may bring about their immunosuppressive action, at least in part, by inhibiting the formation of PGE2. It is paradoxical that corticosteroids inhibit the formation of LXA4 > LTB4, which may explain why steroids interfere with the resolution of inflammation and wound healing. Thus, continued use of corticosteroids results in an EFA deficiency state and leads to decreased formation of LXA4, resolvins, protectins and maresins. This may explain why dexamethasone is beneficial only in mild to moderately severe COVID-19 but not those who are critically ill. The more critically ill with COVID-19 are expected to have an EFA deficiency state and hence are unlikely to respond since they do not have adequate cellular/plasma content of AA/EPA/DHA to form appropriate amounts of PGE2 and LXA4 to resolve inflammation and augment wound healing [117, 133–136]. This may explain why those with COVID-19 who have high levels of cortisol on admission to hospital have a substantially increased risk of dying [215] since they are more likely to have a high degree of EFA deficiency. Hence, if these patients are supplemented with BALs along with other treatment options they are expected to respond favorably.
It is worth noting that glucocorticoids in addition to inhibiting the expression of PLA2, COX-2, LOX, and desaturases enhance the expression of annexin-1, also called lipocortin 1. Annexin-1 shows all the actions of glucocorticoids and induces the anti-inflammatory cytokine IL-10. Annexin-1 suppresses the expression of TNF-α, IL-6, and other cytokines [216]. For the anti-inflammatory action of annexin-1, there is an essential role for LXA4. Both LXA4 and/or annexin-1 seem to enhance the production of IL-10, which suppresses TNF-α action and the consequent tissue injury and lethality [217]. Thus, there is a complex and inter-dependent interaction among PLA2, COX-2, desaturases, annexin-1 and LXA4 in the anti-inflammatory action of corticosteroids.
The recent observation that patients who took statins are likely to have much a reduced mortality rate [218, 219] can be related to their effects on EFA metabolism [220–223]. Statins are known to enhance the formation of AA and LXA4 and thus bring about their beneficial actions. These effects of statins on bioactive lipids are one of the reasons for their anti-inflammatory action. Similarly, the less frequent and mild disease seen in children and decreased incidence of COVID-19 in women can also be ascribed to BALs. For instance, the activity of desaturases decreases with age (Figure 6 B), whereas estrogen stimulates LXA4 synthesis [224, 225]. Thus, it is anticipated that children have a higher capacity to generate GLA, DGLA, AA, EPA and DHA, that can be converted to form beneficial LXA4, resolvins, protectins and maresins to fight SARS-CoV-2. On the other hand, pre-menopausal women generate significantly higher amounts of LXA4 due to the stimulatory action of estrogen and thus are resistant and less likely to develop severe COVID-19. Similarly, even the beneficial action of blocking GM-CSF and administration of colchicine in COVID-19 can be related to their ability to modulate BALs [226–230]. The property of colchicine to potentiate the actions of PGE1 is especially interesting since both PGE1 and LXA4 have similar actions.
A recent study reported that circulating levels of IL-2, IL-4, TNF-α, IFN-γ and C- reactive protein are not associated with severity of COVID-19 symptoms. This indicates that there could be two phases in the pathobiology of COVID-19, one phase characterized by hyperinflammation in the beginning of the disease and another phase of immunosuppression with little or no changes in the pro-inflammatory cytokines as seen in sepsis [231–233]. If this is true, then it will be hazardous to administer dexamethasone, anti-cytokine and other immunosuppressive therapies during the immunosuppressive phase of COVID-19. Hence, one needs to exercise caution in rushing to administer immunosuppressive therapies without measuring plasma cytokines levels and viral load (Figures 8 and 9).
Figure 8
Scheme of events that are likely to occur in SARS-CoV-2 infection and the two phases of cytokine response in COVID-19. Initially there will be hyperinflammation due to release of excess of pro-inflammatory cytokines. Subsequently there could occur diminished release of cytokines and immunosuppression. During the hyperinflammation phase dexamethasone and anti-TNF or anti-IL-6 and other cytokine antagonists will be helpful. Once immunosuppression sets in dexamethasone is not of significant help and inflammation resolution molecules are needed to enhance the recovery process. In severe COVID-19 there will be persistently elevated PGE2 and LXA4 fails to rise to induce resolution of inflammation. In contrast, in mild COVID-19, initially there will be an elevation in PGE2 that falls to the normal physiological level that is accompanied by increase in LXA4 to induce resolution of inflammation as shown in the figure. Compare this with Figure 9. This figure is modified from Das UN. Arch Med Sci 2014; 10: 325-35
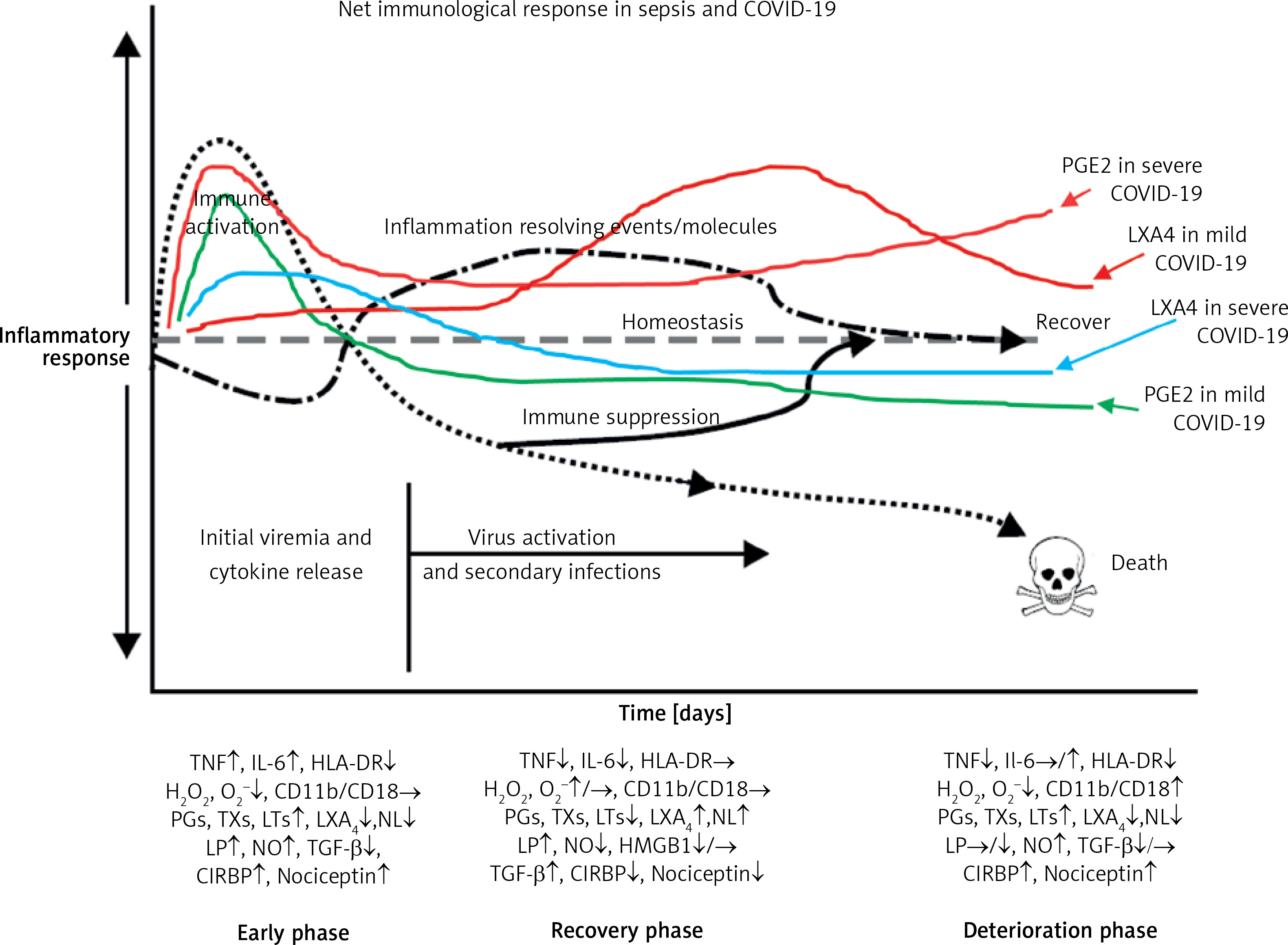
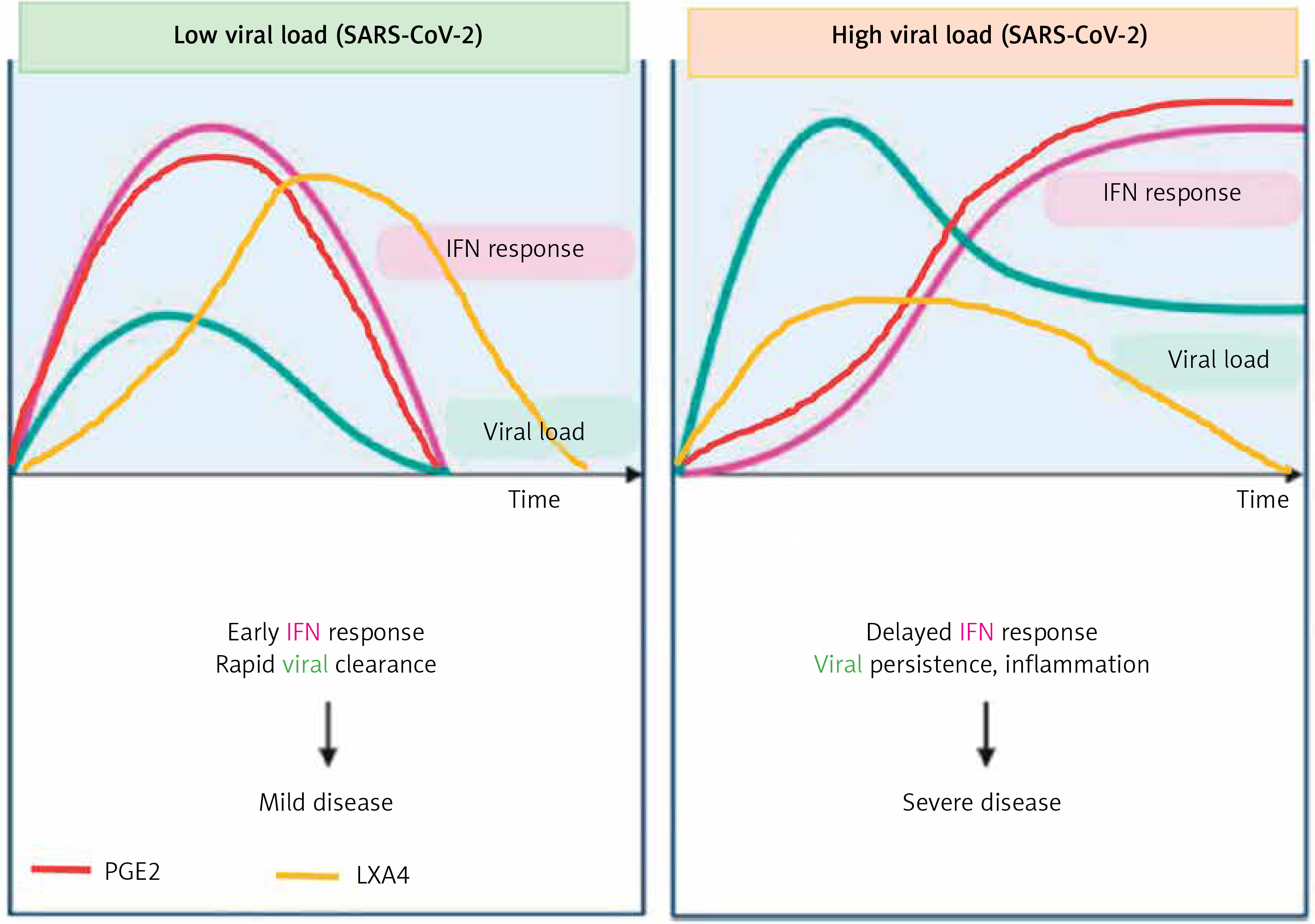
Figure 9
Viral and host factors influence IFN response. When the viral burden is low, IFN production will be adequate to clear the viral infection effectively that is probably accompanied by an initial PGE2 response and timely LXA4 generation to resolve inflammation. When viral load is high, the virus itself may suppress IFN production and stimulate strong PGE2 and weak LXA4 responses, leading to severe COVID-19. The late onset IFN response may aggravate inflammation as seen in severe COVID-19. The PGE2 response may parallel the IFN response. AA release will occur in both mild and severe SARS-CoV-2 infection, but the products formed (PGE2 and LXA4) and their concentrations may differ as shown in the figure. Compare this with Figures 5 and 6
It was reported that H2 blockers such as famotidine could be of significant benefit in those who are admitted with COVID-19 [234, 235]. Those who have taken famotidine were found to have better outcomes. The exact cause for this benefit is not clear. Famotidine could inhibit 3-chymotrypsin-like protease, the enzyme that processes proteins essential for viral replication. It has been noted that famotidine enhances AA formation and thus augments duodenal ulcer healing. Patients with peptic ulcer have low plasma concentrations of AA that reverted to normal following famotidine, fish oil and ARASCO oil (AA rich oil) treatments [236–238]. These results emphasize the importance of AA and other BALs in the pathobiology of peptic ulcer disease and their role in wound healing. It is also significant that Helicobacter pylori, the organism responsible for peptic ulceration, can be inhibited by BALs [239–240], which is in tune with the anti-microbial action of BALs.
Impaired interferon (IFN) type I response in the form of no IFN-β and low IFN-α production and activity was described in those with severe COVID-19 associated with a persistent blood viral load (SARS-CoV-2) and an exacerbated inflammatory response. In these patients, inflammation was found to be driven by upregulation of NF-κB and enhanced production of TNF-α and IL-6 [241, 242]. These data imply that type I IFN deficiency could be a hallmark of severe COVID-19, suggesting that IFN therapy is indicated in these patients. This is supported by the observation that severe COVID-19 is associated with impaired T cell responses in the form of lymphopenia and functional exhaustion of CD4+ and CD8+ T cells, possibly due to deficient IFN production. Furthermore, deficient or dysregulated IFN responses observed in SARS-CoV-2 infection may influence the generation of Treg cells during the recovery phase of COVID-19.
TNF-α and IL-6 enhance PGE2 and thus may indirectly suppress LXA4 concentrations, whereas both PGE2 and LXA4 and their precursor AA decrease TNF-α, IL-6 and IFN production [154, 155, 243–245]. IFN seems to bring about its actions by activating phospholipase C, D and A2 to induce the release of AA (and possibly GLA, DGLA, EPA and DHA) that can be converted to PGE2 and LXA4 [246–248]. PGE2, LXA4, AA, EPA and DHA are potent inhibitors of IFN production [249–254] and IFN can both enhance and decrease PGE2 synthesis depending on the context [255, 256]. These results could be interpreted to imply that if the production of IFN is optimum it leads to synthesis and release of adequate amounts of PGE2 to induce the required degree of inflammation. This optimal action of IFN, PGE2 production and inflammatory reaction results in initiation of the inflammation resolution process so that homeostasis is restored. In the event the levels of IFN are inadequate as seen in severe COVID-19, the production of PGE2 will be suboptimal and so the inflammatory process is not adequate to initiate resolution of inflammation, which results in persistence of inflammation as seen in severe COVID-19. This altered PGE2 production can result in inadequate LXA4 production and failure of wound healing. Furthermore, both PGE2 and LXA4 inhibit IFN and TNF-α production, whereas IFN and TNF-α are inducers of PGE2 production. Similar interaction between EPA/DHA and their eicosanoids is expected for IFN and TNF-α. IFN (IFN-γ but not IFN-α and IFN-β) can enhance the production of TNF-α [257–259]. This positive and negative feedback among AA/EPA/DHA/GLA/DGLA/PGE2, LXA4, resolvins, protectins and maresins and IFN and TNF-α is needed to effectively eliminate SARS-CoV-2 and other microbes and eventually restore homeostasis. Thus, inadequate production of IFN-α in those with COVID-19 (that may in part be due to inadequate tissue content of GLA, DGLA, AA, EPA, DHA) leads to relatively low (but more than normal) levels of PGE2 [260] and excess of LXA4 (this ratio between PGE2 and LXA4 is relative). As a result of this relative deficiency of PGE2, excess production of TNF-α and IL-6 occurs, leading to the cytokine storm seen in severe COVID-19. Excess TNF-α causes decreased formation of AA/EPA/DHA due to its inhibitory action on desaturases [137]. This further aggravates deficiency of AA/EPA/DHA that can result in persistent excess production of TNF-α and IL-6 due to the absence of negative feedback regulation of these fatty acids on cytokines. In this scenario, administration of GLA/DGLA/AA/EPA/DHA may be of significant benefit to suppress excess production of TNF-α and IL-6 and restore LXA4 levels to normal and halt this vicious circle of events. This implies that use of IFN alone will not be of benefit in COVID-19 unless it is co-administered with BALs.
This imbalance in the immune response, cytokines and BALs is supported by the observation that in those with severe COVID-19 the germinal centers (where B and T cells are formed to meet the demands of infections for an adequate immune response) in the thoracic lymph nodes and spleens are absent, which seems to be associated with high plasma levels of TNF-α, implying that this cytokine is suppressing the immune system [261, 262]. These results are interesting in the light of the observation that 15-PGDH deficient animals not only have a twofold increase in bone marrow, colon, and liver PGE2 levels but also showed augmented hematopoietic capacity [185]. This implies that AA/EPA/DHA, IFN, TNF-α and PGE2 dysregulation results in an inadequate immune response in COVID-19 (Figure 10).
Figure 10
Scheme showing interaction among SARS-CoV-2, host cells, immunocytes, AA and cytokines. Similar interaction may exist among SARS-CoV-2, host cells/immunocytes, EPA/DHA and resolvins, protectins and maresins
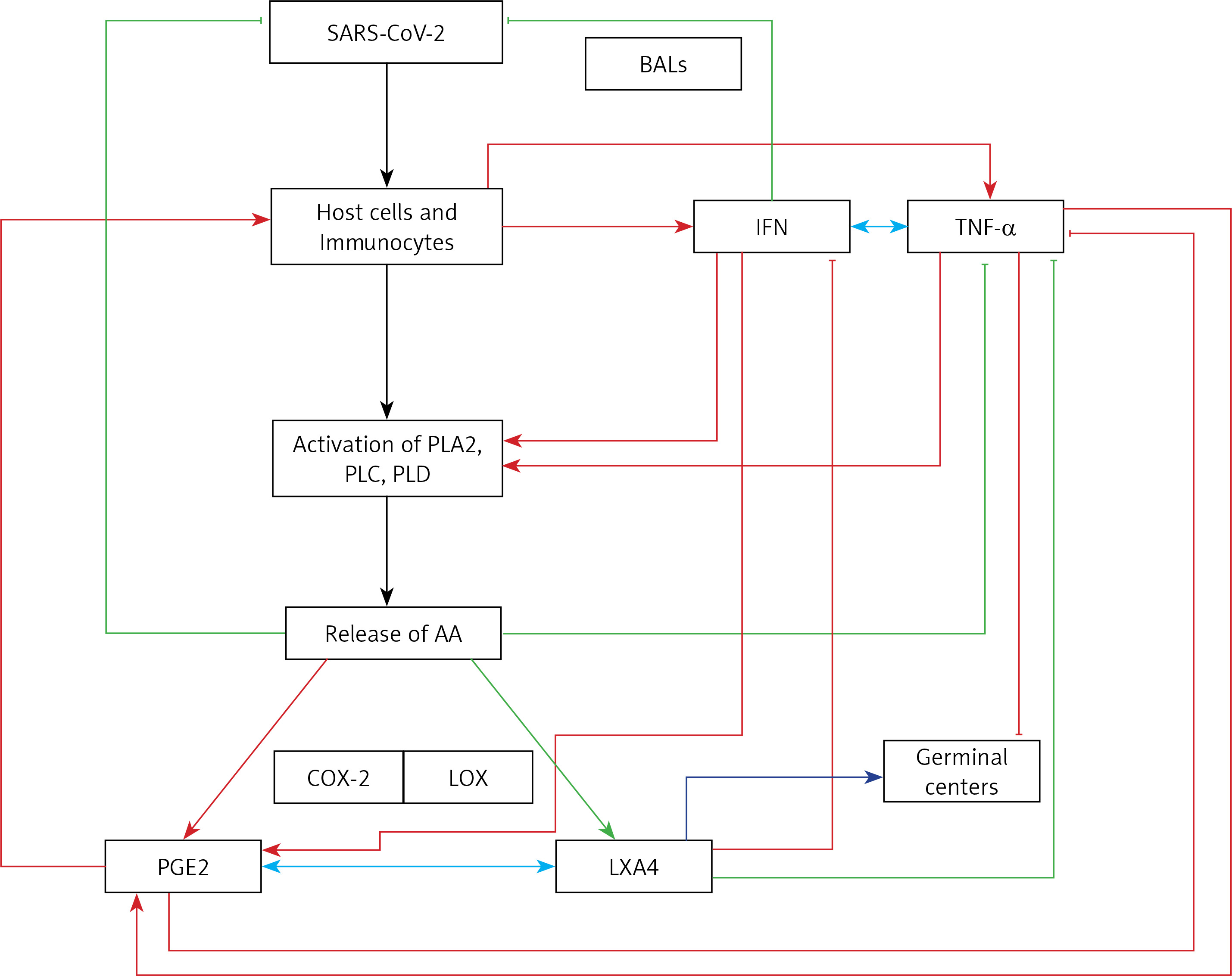
The recent report that bradykinin may have a critical role in the cytokine storm seen in COVID-19 [263] can also be explained in terms of BALs. For instance, it is known that GLA, EPA and DHA (and possibly AA, PGE2, LXA4) can alter cell membrane fluidity and thus alter the binding characteristics of bradykinin receptor (B2R) to bradykinin [264]. In addition, bradykinin stimulates PLA2 and enhances PGE2 formation [265–267]. PGE2 restrains mast cell activity and LXA4 can resolve inflammation induced by mast cell degranulation [268–270]. These results imply that the actions of bradykinin can be regulated and inhibited by LXA4 and other similar compounds – resolvins, protectins and maresins (Figure 11). Based on the concepts proposed here, we successfully treated 10 consecutive COVID-19 patients by the administration of oral BALs, oral and inhaled corticosteroids and heparin, oral B1, B6, B12 and vitamin C (that form co-factors of EFA metabolism), and insulin (for hyperglycemia and incidentally it has anti-inflammatory action and enhances the activities of desaturases). In this open label study, 5 were asymptomatic, 3 had mild disease and 2 had moderately severe disease with co-morbid conditions – type 2 diabetes mellitus, hypertension, and coronary heart disease. All recovered completely without any side effects. In addition, these patients were on statins, which are known to be of benefit in COVID-19 [271, 272].
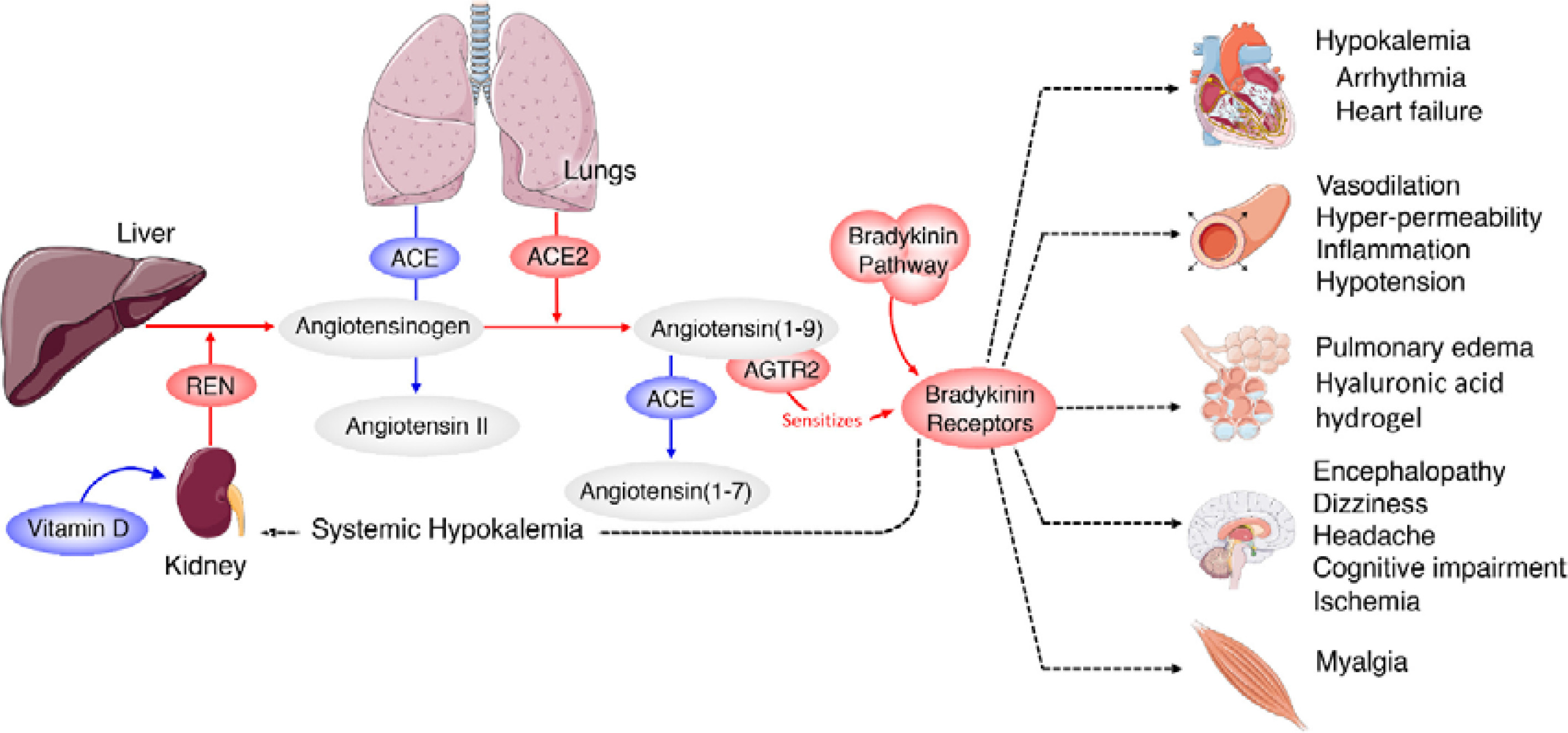
Future research strategies and therapeutic implications
The concepts presented here can be verified by studying the effect of BALs (especially AA, EPA, DHA, LXA4, resolvins, protectin and maresins) on the survival and proliferation of SARS-CoV-2, SARS, and MERS in vitro and in vivo. It is predicted that BALs, especially LA and AA, inactivate SARS-CoV-2, SARS, and MERS. Studies need to be performed to know whether PUFAs and their metabolites can influence the plasticity of macrophages to form M1 and M2 phenotypes. It is important to evaluate the role of desaturases, COX-2 and 5-, 12-, and 15-LOX enzymes and different types of phospholipases in COVID-19. It is predicted that those who have severe COVID-19 will show exaggerated expression of PLA2 and COX-2, whereas those who recover from COVID-19 are likely to show both COX-2 and 12-LOX and 15-LOX expression resulting in synthesis and release of adequate amounts of LXA4 to result in resolution of inflammation (Figures 5–8). This can be confirmed by measuring plasma levels of various BALs that will throw light on whether a deficiency of LXA4, resolvins, protectins and maresins could be responsible for the severe COVID-19 and high degree of mortality. It is predicted that timely production of PGE2 and LXA4 may fail to occur in these patients (Figures 5–8). Similarly, a close association between the plasma levels of IL-6, TNF-α and IFN and various BALs could be seen that could be correlated with the clinical picture.
Based on the evidence presented here, it is suggested that LA, GLA, DGLA, AA, EPA and DHA could be administered to prevent and manage COVID-19. Of all the BALs, it is likely that LA and AA may be of significant benefit in the treatment of COVID-19. The clinical implications of potential use of BALs in the prevention and treatment of COVID-19 lies in the fact that the antibodies developed against SARS-CoV-2 (due to either infection or immunization) may not last long enough to prevent a second infection. Alternatively, SARS-CoV-2 may undergo substantial mutation(s) such that the initial immunization against the current strain(s) may not work against the mutated viruses. In such a scenario, it is worthwhile to investigate whether a combination of vaccines against SARS-CoV-2 and BALs (especially LA and AA) will be of significant benefit in preventing COVID-19. Since BALs have the broad property of inactivating all types of enveloped viruses, it is likely that their (BALs, especially AA) administration may work in the prevention and treatment of all types of SAR viruses irrespective of changes in their genome.