Introduction
Oral squamous cell carcinoma (OSCC) is the most frequent neoplasm of the oral cavity [1]. Risk factors, mainly including alcohol consumption, tobacco use, betel nut chewing and human papillomavirus (HPV) infection, may be responsible for the etiology of OSCC [2, 3]. Surgery is the primary treatment method for patients with early OSCC [4]. Combined with surgery, adjuvant radiotherapy and chemotherapy are widely used to treat late-stage OSCC, but the overall survival rate remains at a low level partly due to the development of resistance to radiation therapy or chemotherapy [5]. Gefitinib, an inhibitor of EGFR and HER-2 kinases, has potent antitumor activity in a variety of human cancers such as lung, breast and head and neck cancers [6]. However, the generated resistance of gefitinib treatment obviously reduces the therapeutic effect [7–9]. Therefore, exploring the molecular mechanisms of gefitinib resistance acquisition in OSCC is critical for developing effective treatment methods.
Bone marrow stromal cell antigen 2 (BST2), also known as HM1.24, CD317 or tetherin, is a type II transmembrane glycoprotein that inhibits the release of various viruses including HIV-1, ebola and Lassa viruses [10–12]. BST2 is found to be expressed in bone marrow stromal cells and terminally differentiated human B cells and is relevant to pre-B cell growth by enhancing cell-cell interaction [13, 14]. Additionally, accumulating evidence suggests that BST2 was aberrantly expressed in various tumors such as nasopharyngeal cancer, gastric cancer and oral cavity cancer [11, 13, 15]. Moreover, BST2 is associated with tumor invasion, progression or drug-resistant phenotypes [16, 17]. However, the effect of BST2 on OSCC is not well described.
In the present study, we determined the expression of BST2 in OSCC tissues, and detected the effects of BST2 on the growth and gefitinib resistance in OSCC cells, and the underlying mechanism was also analyzed. Multiple biological experiments demonstrated that BST2 could promote growth and induce gefitinib resistance in OSCC cells through regulating the EGFR pathway.
Material and methods
Clinical specimens
Sixty pairs of OSCC tissues and the corresponding non-tumor tissues were acquired from previously untreated OSCC patients who underwent surgery at Shanghai Songjiang District Central Hospital. Informed consent was obtained from all patients before the experiment. This study was approved by the Human Research Scientific Ethics Committee of Shanghai Songjiang District Central Hospital.
Cell culture and transient transfection
OSCC cell lines (H157, HSC-2) were purchased from Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin under a humidified incubator of 5% CO2 at 37°C.
Full length BST2 cDNA was amplified by PCR and inserted into the pcDNA3.1 vector to obtain an overexpressing BST2 plasmid (pcDNA3.1-BST2). Two different small interference RNAs (siRNA) targeting BST2 (siBST2-1#, siBST2-2#) and scrambled siRNA control (NC) were purchased from GenePharma Co., Ltd (Shanghai, China). Cells were transfected with plasmids or siRNAs using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. After 24 h of incubation, the cells were collected for further analysis.
Quantitative reverse transcription-PCR (qRT-PCR) analysis
Total RNAs from tissues and cells were extracted using TRIzol reagent (Invitrogen) and reverse transcribed with the Reverse Transcriptase Kit (Takara, Dalian, China) to obtain the cDNAs. qRT-PCR was performed with SYBR Premix Ex taq (Takara Bio, Shiga, Japan) and analyzed on an ABI 7500 Fast Real-Time PCR system. The primer sequences were as follows: BST2, forward primer 5′-GGAGGAGCTTGAGGGAGAG-3′ and reverse primer 5′-CTCAGTCGCTCCACCTCTG-3′; β-actin, forward primer 5′-AGCCTCGCCTTTGCCGA-3′ and reverse primer 5′-CTGGTGCCTGGGGCG-3′. For normalization of qRT-PCR analysis, β-actin was used as an internal control. The relative levels of mRNA were calculated by the 2–ΔΔCt method.
Western blot
Total proteins from tissues and cells lysed with RIPA lysis buffer containing a protease inhibitor (Sigma-Aldrich) were equally separated by SDS-PAGE and transfected to PVDF membranes. The membranes were blocked with 5% fat-free milk for 2 h at room temperature and incubated with primary antibodies against BST2, p21, cyclin A, cyclin D1, GAPDH, Bcl-2, Bax, cleaved caspase-3, p-EGFR, EGFR, Erk, p-Erk, Akt, p-Akt, ABCG2 and BCRP at 4°C overnight, following by culturing with HRP-conjugated secondary antibodies for 2 h at room temperature. The western blots were photographed by the ECL detection system (Amersham Biosciences, Piscataway, NJ, USA), and quantified by ImageJ software (v1.8.0, National Institutes of Health, Maryland, USA).
Immunohistochemical analysis
Paraffin sections (4 μm thick) from the tissues were deparaffinized in xylene solution, rehydrated with graded alcohols, blocked with hydrogen peroxide and subjected to antigen recovery treatment with citrate buffer (pH 8.0) by heating for 20 min. Then 0.5% H2O2 in methanol was used to block the endogenous peroxidase activity. The sections were incubated with a primary antibody against BST2 overnight at 4°C. Subsequently, the sections were incubated with an HRP-conjugated secondary antibody. After washing with PBS, the sections were processed with the Betazoid DAB Chromogen Kit (Biocare), and then counterstained with hematoxylin. The samples were observed under a light microscope (Olympus Corporation, Japan).
Cell proliferation assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to determine the cell proliferation. Twenty-four h after transfection, the transfected cells were seeded in 96-well plates and then incubated for 24, 48, 72 and 96 h, and MTT solution was then added into each well. The optical density (OD) at 490 nm was measured by a microplate reader. To analyze the effects of BST2 on gefitinib resistance, 24 h after transfection, the transfected cells were exposed to different concentrations of gefitinib for 72 h, MTT was then added and the OD value at 490 nm was tested by a microplate reader. Half maximal inhibitory concentration (IC50) was assessed by GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA).
Analysis of cell cycle and apoptosis by flow cytometry
Cells were transfected with pcDNA3.1-BST2, BST2 overexpression control group (pcDNA3.1), siBST2-1#, siBST2-2#, siRNA control (NC) for 24 h, and then harvested for further analysis. To analyze the effects of BST2 on gefitinib resistance, cells transfected with pcDNA3.1-BST2 were exposed to 0.1 μM gefitinib for 24 h, followed by collecting the cells for further analysis.
For apoptosis analysis, the harvested cells were resuspended in binding buffer and stained with FITC-Annexin-V/PI. A FACScan flow cytometer (BD Biosciences, Franklin lakes, NJ, USA) was applied to determine the cell apoptotic rates.
For cell cycle analysis, the cycle of the harvested cells was detected by Cell Cycle Detection Kits (KeyGEN Biotech) based on the manufacturer’s instructions, measured by the FACSVerse flow cytometer (BD Biosciences) and analyzed by FlowJo software (Tree Star, Stanford, CA).
Statistical analysis
All experiments were repeated at least 3 times. The data were presented as mean ± standard deviation (SD) and analyzed by SPSS 16.0 software (SPSS, Chicago, IL, USA). Differences between two or more groups were determined by Student’s t-test. Differences with p < 0.05 were considered statistically significant.
Results
BST2 expression was up-regulated in OSCC tissues
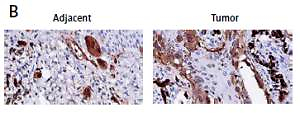
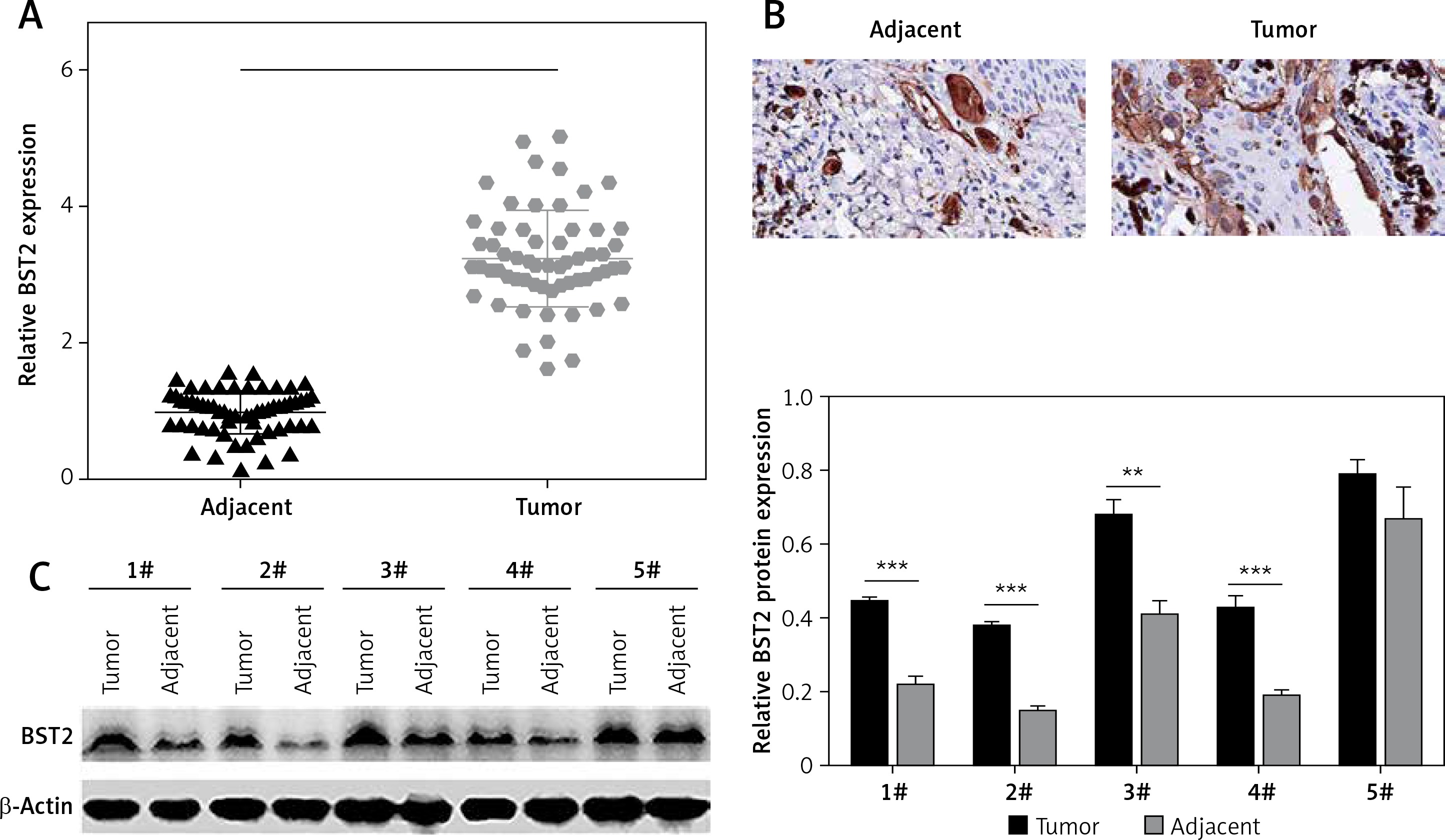
To explore the role of BST2 in OSCC, we determined the expression of BST2 in OSCC tissues using qRT-PCR, immunohistochemistry and western blot, respectively. As shown in Figure 1 A, qRT-PCR showed that BST2 had a higher expression level in OSCC tissues than that in the adjacent normal tissues. Immunohistochemistry indicated that the positive staining of BST2 in OSCC tissue sections showed an obvious increase compared with the normal tissues (Figure 1 B). The protein expression of BST2 in five randomly selected OSCC tissues was significantly up-regulated as compared to the adjacent normal tissues (Figure 1 C). These results suggested that both mRNA and protein expression of BST2 was up-regulated in OSCC tissues.
Figure 1
Expression of BST2 in OSCC tissues. A – Relative mRNA expression of BST2 in OSCC tissues was determined by qRT-PCR. B – Positive staining of BST2 in OSCC tissues was measured by immunohistochemistry. C – Protein expression of BST2 in five randomly selected OSCC tissues was detected by western blot. Student’s t test was used for statistical analysis
***p < 0.01; **p < 0.05 versus the adjacent normal tissues.
BST2 overexpression enhanced OSCC cell proliferation
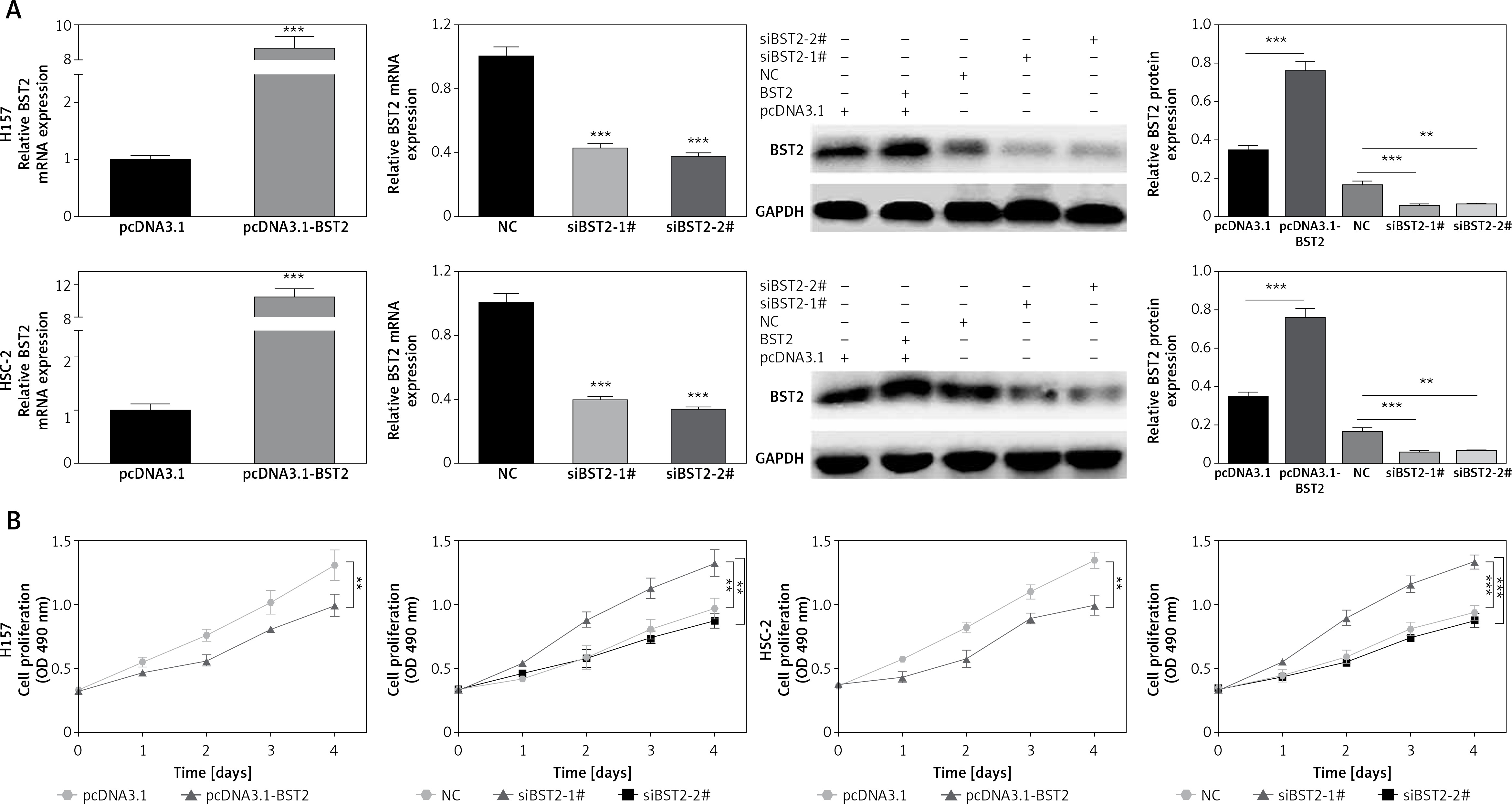
OSCC cell lines H157 and HSC-2 were separately overexpressed and knocked down by transfecting with BST2 overexpression vector and siBST2, respectively. Firstly, the transfection efficiency was determined by qRT-PCR and western blot. As shown in Figure 2 A, BST2 plasmids (pcDNA3.1-BST2) effectively increased the mRNA and protein expression of BST2 in H157 and HSC-2 cell lines compared with the control group (pcDNA3.1). Two types of siRNA for BST2 (siBST2-1#, siBST2-2#) significantly decreased the expression of BST2 in both cell lines compared to the control (NC). To investigate the effect of BST2 on OSCC cell viability, MTT assay was used. The results revealed that BST2 overexpression promoted the proliferation of OSCC cells, while knockdown of BST2 remarkably inhibited the cellular proliferation compared with the control (Figure 2 B).
Figure 2
Effect of BST2 on proliferation of OSCC cells. A – After H157 and HSC-2 cell lines were transfected with pcDNA3.1-BST2, BST2 expression control group (pcDNA3.1), siBST2-1#, siBST2-2# or siRNA control (NC), the transfection efficiency was assessed by qRT-PCR and western blot respectively. B – The proliferation of OSCC cells was determined by MTT assay. Student’s t test was used for statistical analysis
***p < 0.01; **p < 0.05 versus control.
BST2 regulated OSCC cell cycle progression
To investigate the effect of BST2 on the OSCC cell cycle, the OSCC cell cycle was evaluated by flow cytometry. 24 h after transfecting H157 and HSC-2 cells with pcDNA3.1, pcDNA3.1-BST2, siBST2-1#, siBST2-2# or NC, flow cytometry indicated that the percentage of cells transfected with pcDNA3.1-BST2 in the G1 phase was decreased, whereas BST2 knockdown increased the percentage of cells in the G1 phase as compared to the corresponding control group (Figure 3 A).
Figure 3
Effect of BST2 on OSCC cell cycle progression. After H157 and HSC-2 cell lines were transfected with pcDNA3.1-BST2, BST2 expression control group (pcDNA3.1), siBST2-1#, siBST2-2# or siRNA control (NC), A – the cell cycle progression was measured by flow cytometry; B – expression of p21, cyclin A and cyclin D1 was evaluated by western blot. Student’s t test was used for statistical analysis
***p < 0.01; **p < 0.05 versus control.
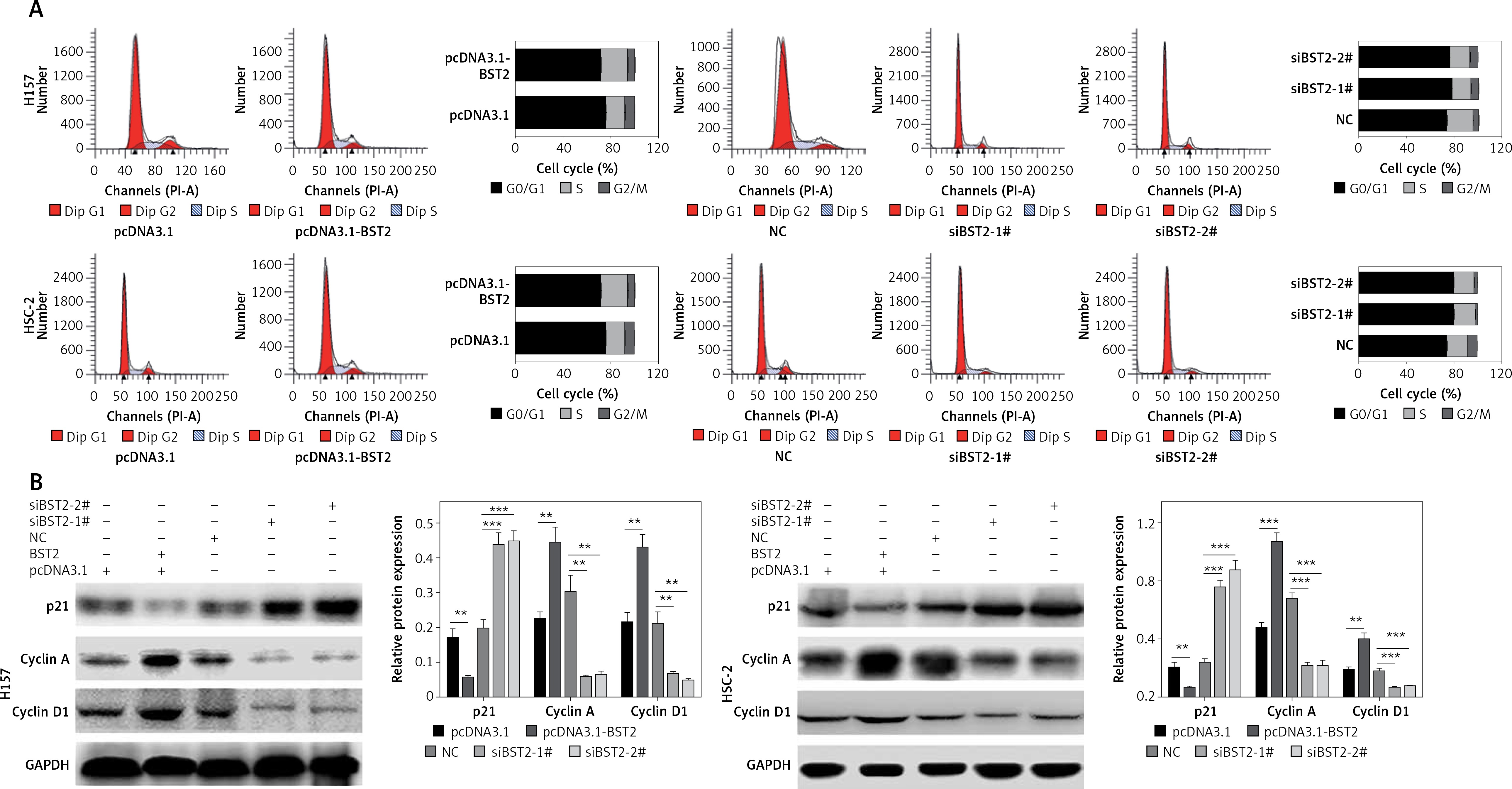
As shown in Figure 3 B, BST2 overexpression up-regulated the expression of cyclin A and cyclin D1 and down-regulated p21 expression in H157 and HSC-2 cells as compared with the control group. However, down-regulation of cyclin A and cyclin D1 expression and up-regulation of p21 expression were observed in H157 and HSC-2 cells transfected with siRNAs BST2. It is known that cyclin A and cyclin D1 are key proteins in the regulation of cell cycle progression, and p21 is involved in DNA damage-induced cell cycle arrest and blocking DNA replication and repair [18].
BST2 overexpression inhibited OSCC cell apoptosis
The effect of BST2 on OSCC cell apoptosis was also detected after transfection for 24 h. Images of flow cytometry and the quantification of the result showed that the BST2 overexpression greatly decreased the apoptosis of H157 and HSC-2 cells, and knockdown of BST2 significantly increased the cell apoptosis as compared to the corresponding controls (Figure 4 A).
Figure 4
Effect of BST2 on apoptosis of OSCC cells. After H157 and HSC-2 cell lines were transfected with pcDNA3.1-BST2, BST2 expression control group (pcDNA3.1), siBST2-1#, siBST2-2# or siRNA control (NC), A – apoptosis of OSCC cells was detected by flow cytometry and the quantification of apoptosis was calculated and listed on the left side; B – protein expression of Bcl-2, Bax and cleaved caspase-3 was determined by western blot. Student’s t test was used for statistical analysis
***p < 0.01; **p < 0.05 versus control.
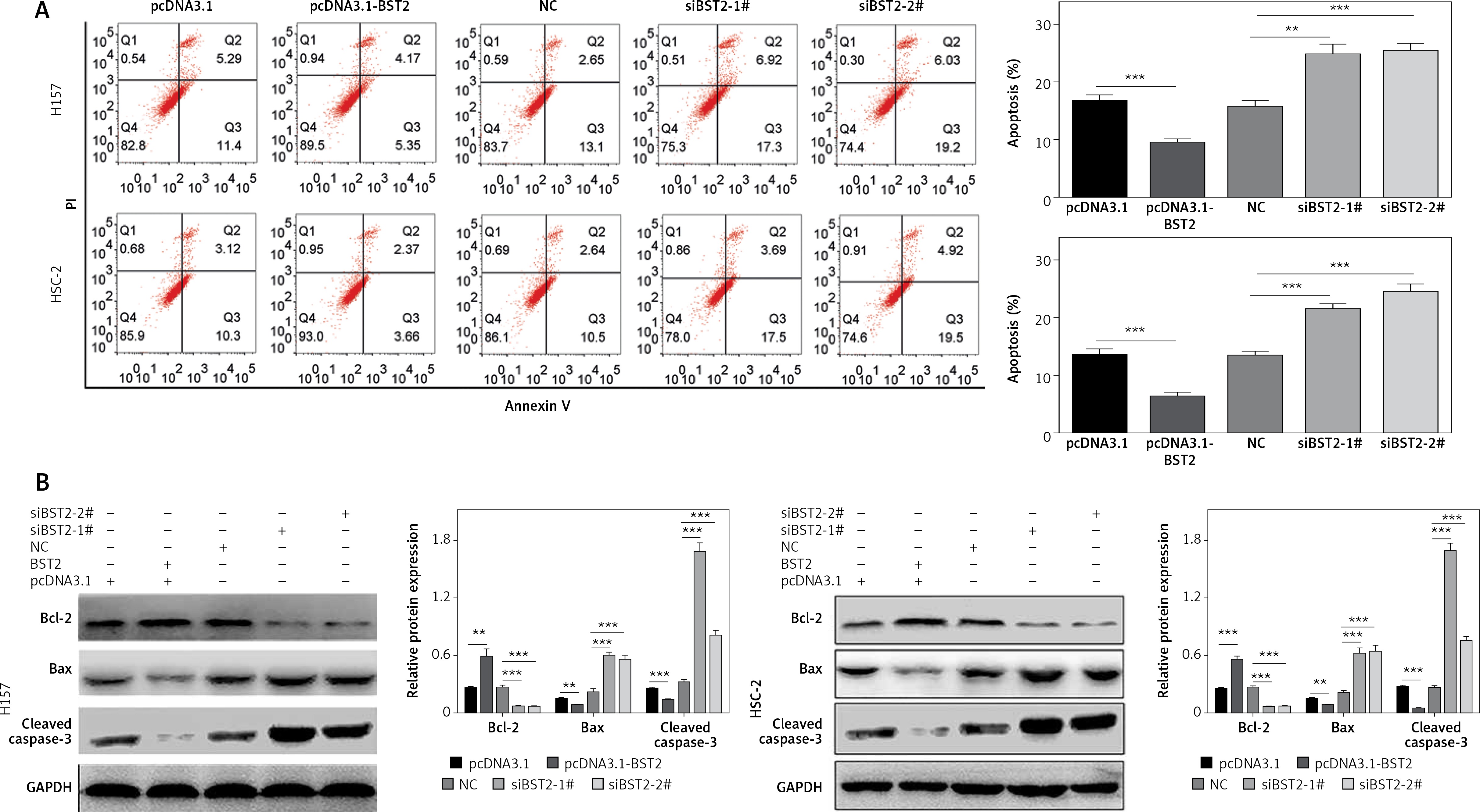
Western blot further confirmed that the expression of BCL-2, an anti-apoptosis protein, was up-regulated by BST2 overexpression and down-regulated by BST2 knockdown. Indeed, pro-apoptosis proteins BAX and active caspase-3 expression levels were down-regulated by BST2 overexpression and up-regulated by BST2 knockdown (Figure 4 B). Collectively, BST2 could regulate proliferation, cycle and apoptosis of OSCC cells.
BST2 overexpression induced gefitinib resistance in OSCC cells
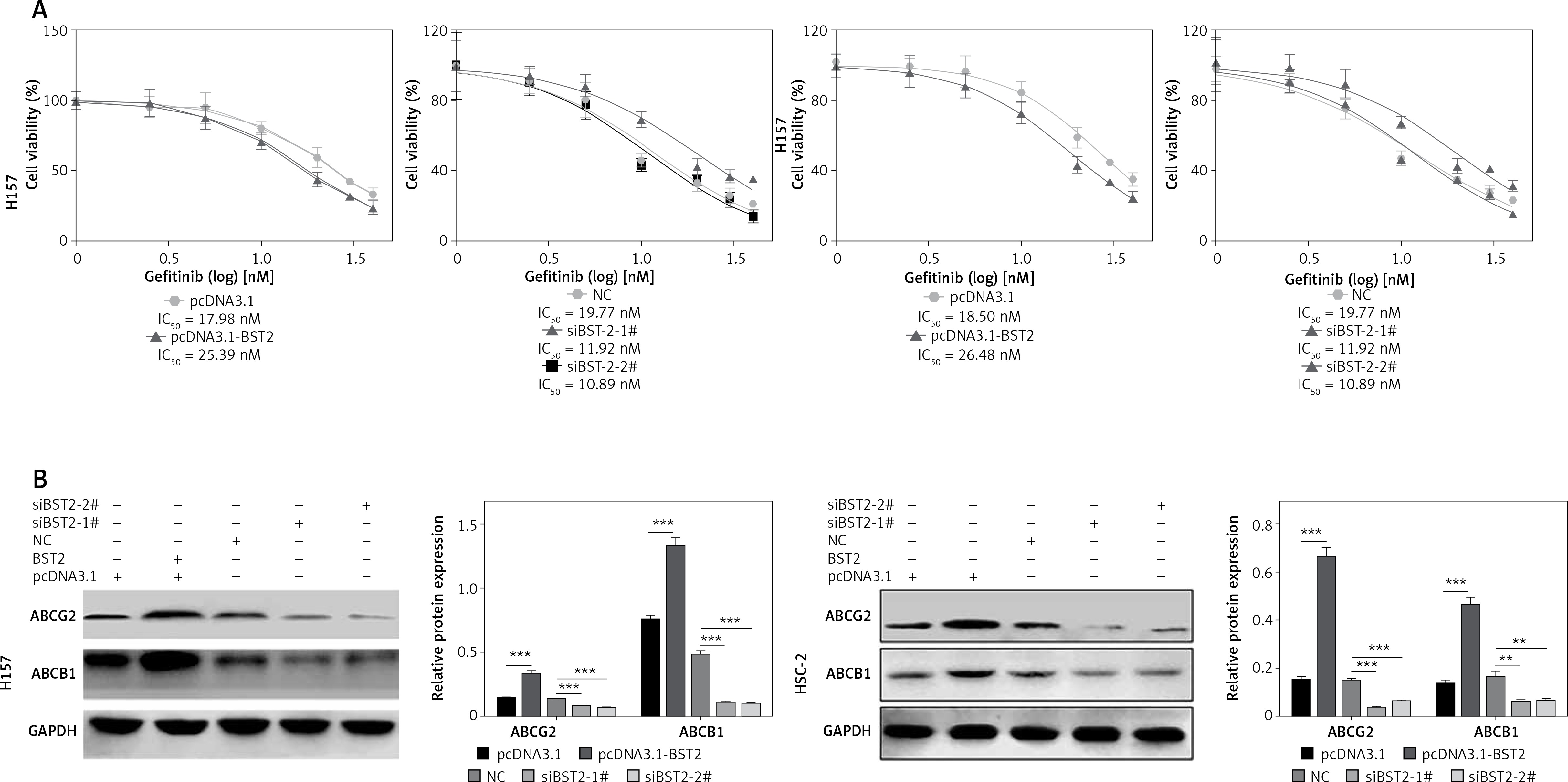
H157 and HSC-2 cells transfected with pcDNA3.1, pcDNA3.1-BST2, siBST2-1#, siBST2-2# or NC were exposed to different concentrations of gefitinib for 72 h, and determined by MTT assay. As shown in Figure 5 A, the IC50 of gefitinib was increased in H157 and HSC-2 cells with BST2 overexpression while knockdown of BST2 decreased the IC50 of gefitinib as compared to the corresponding control groups. Western blot indicated that the expression levels of the drug resistance-related proteins ABCG2 and BCRP in H157 and HSC-2 cells were enhanced by BST2 overexpression, whereas knockdown of BST2 reduced the expression of ABCG2 and BCRP corresponding control groups (Figure 5 B). Taken together, BST2 overexpression induced gefitinib resistance in OSCC cells.
Figure 5
Effect of BST2 on gefitinib resistance. After H157 and HSC-2 cell lines were transfected with pcDNA3.1-BST2, BST2 expression control group (pcDNA3.1), siBST2-1#, siBST2-2# or siRNA control (NC), A – the transfected cells were treated with different concentrations of gefitinib (0, 0.5, 0.75, 1, 1.25, 1.5 nM) and then determined by MTT assay to obtain the IC50 of gefitinib; B – expression levels of the drug resistance proteins ABCG2 and BCRP in H157 and HSC-2 cells were measured by western blot. Student’s t test was used for statistical analysis
***p < 0.01; **p < 0.05 versus control.
BST2 overexpression activated the epidermal growth factor receptor (EGFR) pathway in OSCC cells
To further explore the molecular mechanisms of BST2 in the cell cycle, apoptosis and gefitinib resistance, H157 and HSC-2 cells with BST2 overexpression were treated with or without gefitinib, followed by western blot and flow cytometry analysis. Compared with the control group, the phosphorylation levels of EGFR, Erk and Akt were increased by BST2 overexpression, whereas the high expression was reversed by gefitinib treatment (Figure 6 A). Additonally, the expression of EGFR, Erk and Akt had no significant difference among three groups. These results indicated that BST2 overexpression significantly activated the EGFR pathway.
Figure 6
Effect of BST2 on the EGFR signaling pathway. After H157 and HSC-2 cells with the transfection of BST2 were treated with or without gefitinib, A – the expression levels of p-EGFR, EGFR, p-Erk, Erk, p-Akt, and Akt in the EGFR pathway were assessed by western blot. The cell cycle (B) and apoptosis (C) were detected by flow cytometry. Student’s t test was used for statistical analysis
***p < 0.01; **p < 0.05 versus control.
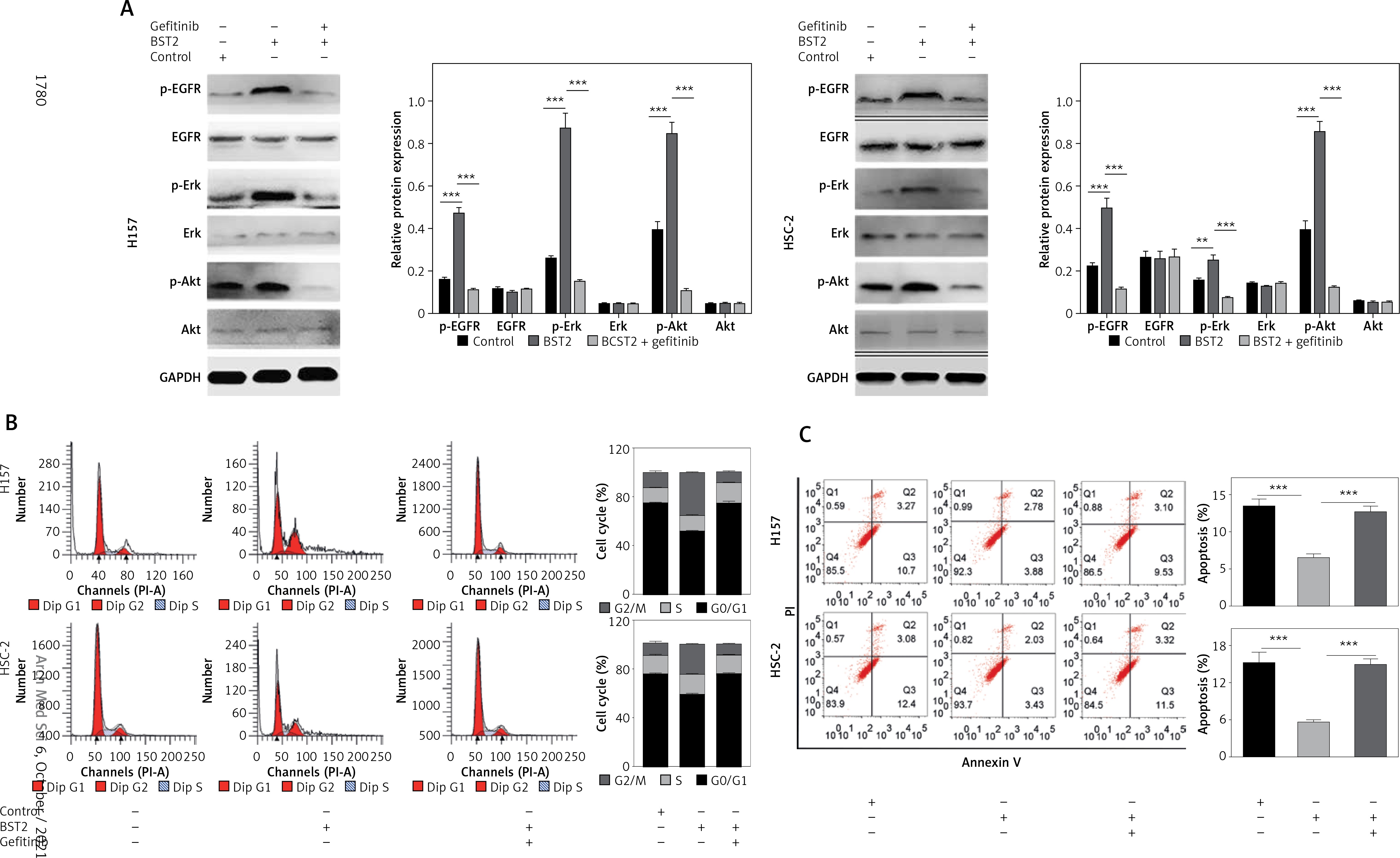
Since the EGFR pathway plays an important role in the cell cycle and apoptosis [19], the cells were also determined by flow cytometry. As shown in Figure 6 B, the percentage of H157 and HSC-2 cells in the G1 phase was decreased by BST2 overexpression compared to the control group. However, the gefitinib treatment up-regulated the percentages of H157 and HSC-2 cells in the G1 phase. Additionally, the inhibition of OSCC cell apoptosis caused by BST2 overexpression was markedly altered with gefitinib treatment (Figure 6 C). Collectively, the effect of BST2 on OSCC cells might be through activating the EGFR signaling pathway.
Discussion
Here, BST2 was found up-regulated in OSCC tissues. BST2 overexpression promoted OSCC cell proliferation, regulated the cell cycle progression and inhibited the apoptosis of OSCC cells. Consistently, BST2 has been reported to be involved in the invasion and proliferation of various tumors such as OSCC [15], indicating its important role in OSCC progression. OSCC’s standard therapeutic method is a combination of surgery, radiotherapy and chemotherapy [1]. However, the survival rate of the advanced OSCC patients is still low, which results from many factors including tumor metastasis, invasion and particularly chemotherapy resistance [1, 5]. Thus, exploring the molecular mechanisms of chemotherapy resistance acquisition in OSCC is a potential research direction. In this study, BST2 overexpression contributed to gefitinib resistance in OSCC cells. Similarly, BST2 was reported to confer chemotherapy resistance in other cancers [11, 16].
The molecular mechanisms of BST2 in the proliferation and chemotherapy resistance in OSCC cells were further explored. The MTT assay showed that BST2 overexpression enhanced the proliferation of OSCC cells. Flow cytometry indicated that the percentage of OSCC cells in the G1 phase was decreased by BST2 overexpression. Also, western blot revealed that BST2 over-expression in OSCC cells significantly increased the expression of cyclin A and cyclin D1 and decreased p21 expression as compared to the control group. Cyclin A and cyclin D1 are members of the cyclin family that is essential for regulating progression through the cell cycle [18]. P21, a potent cyclin-dependent kinase inhibitor, is considered as a checkpoint regulator for cell cycle progression at G1 and S phase through inhibiting the activity of cyclin-CDK4/6, -CDK2, and -CDK1 complexes [19, 20]. Taken together, BST2 promoted OSCC cell cycle progression through regulating the levels of cyclin A, cyclin D1 and p21. Additionally, flow cytometry analysis also indicated that BST2 overexpression inhibited the apoptosis of OSCC cells, which was accompanied by enhancing anti-apoptosis protein BCL-2 expression and reducing pro-apoptosis proteins BAX and active caspase-3 expression levels [21]. Collectively, these results suggested that BST2 could regulate the cell proliferation, cycle progression and apoptosis through regulating apoptosis-related proteins.
The antitumor activity of gefitinib has been demonstrated in many cancers such as lung and head and neck cancers [6, 7]. However, previous evidence also suggested that patients could ultimately acquire gefitinib resistance [8, 9, 22]. A previous study suggested that gefitinib inhibits the phosphorylation of EGFR and its downstream signaling pathways through binding to the catalytic kinase domain of EGFR, eventually leading to enhanced cell apoptosis and suppressed cell proliferation, adhesion, migration and survival [23, 24]. In the present study, we found that BST2 overexpression significantly enhanced the gefitinib resistance in OSCC cells, and knockdown of BST2 considerably reduced the gefitinib resistance. Moreover, our results indicated that BST2 overexpression-induced activation of the EGFR pathway was obviously reversed after gefitinib treatment. Based on the evidence that activation of EGFR signaling could promote cell proliferation, migration, and cell cycle progression and inhibit cell apoptosis [19, 25, 26] and the EGFR signaling pathway is closely related to gefitinib resistance [27, 28], we concluded that the effect of BST2 on OSCC cell growth and gefitinib resistance might be through regulating the EGFR signaling pathway.
In conclusion, BST2 was highly expressed in OSCC tissues compared with the normal tissues. Our results also showed that the critical role of BST2 in proliferation, cycle progression, apoptosis and gefitinib resistance of OSCC cells was associated with activation of the EGFR signaling pathway. Therefore, BST2 might be used as a potent therapeutic target to alter chemotherapy resistance in OSCC. Certainly, this is a preliminary study revealing the involvement of BST2 in OSCC progression and there is a lack of animal experiments and clinical trials, which are limitations of this study. More comprehensive basic research needs to be performed to further verify other involved underlying mechanisms of BST2 in inducing gefitinib resistance.