A 71-year-old Chinese woman, who received the first dose of COVID-19 vaccination on 1 July 2021 and a second dose on 27 July 2021, was admitted to the Department of Gastroenterology on 2 February 2022 due to intractable vomiting with abdominal pain for approximately 20 days. The vomiting symptom gradually worsened, accompanied by new symptoms such as dizziness, unstable walking, dysarthria, and malaise. Previously, she had suffered from repeated vomiting and abdominal pain, on 29 July 2021 and in October, respectively. Brain DWI revealed high-intensity signal in the brain stem and right cerebellum (Figure 1 B). She was considered as a posterior circulation infarction and referred to a neurologist.
Figure 1
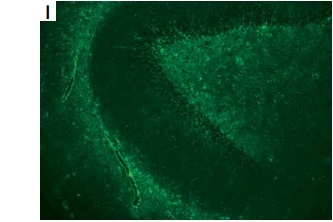
A – Timeline of the patient. B – Brain DWI revealed high-intensity signal in the brain stem and right cerebellum. C–E – Brian T2-flaire hyperintense signal in right former cornu of ventricle, temporal horn, pons, medulla oblongata, and cerebellum lesions. F–H – Spine T2-flaire hyperintensity in the cervical cord and thoracic cord from 4th to 8th level. I, J – Representative positive signal of GFAP morphological characteristic, rather than AQP4, from neuroglia was found in hippocampus and cerebellum, detected using monkey cerebellar tissue-based assay. K – Haematoxylin and eosin staining showed the cells with thyroid morphology in teratoma section of the patient. L, M – Images of immunohistochemical staining demonstrated the negative of antibodies against GFAP and AQP4 (magnification 400×)
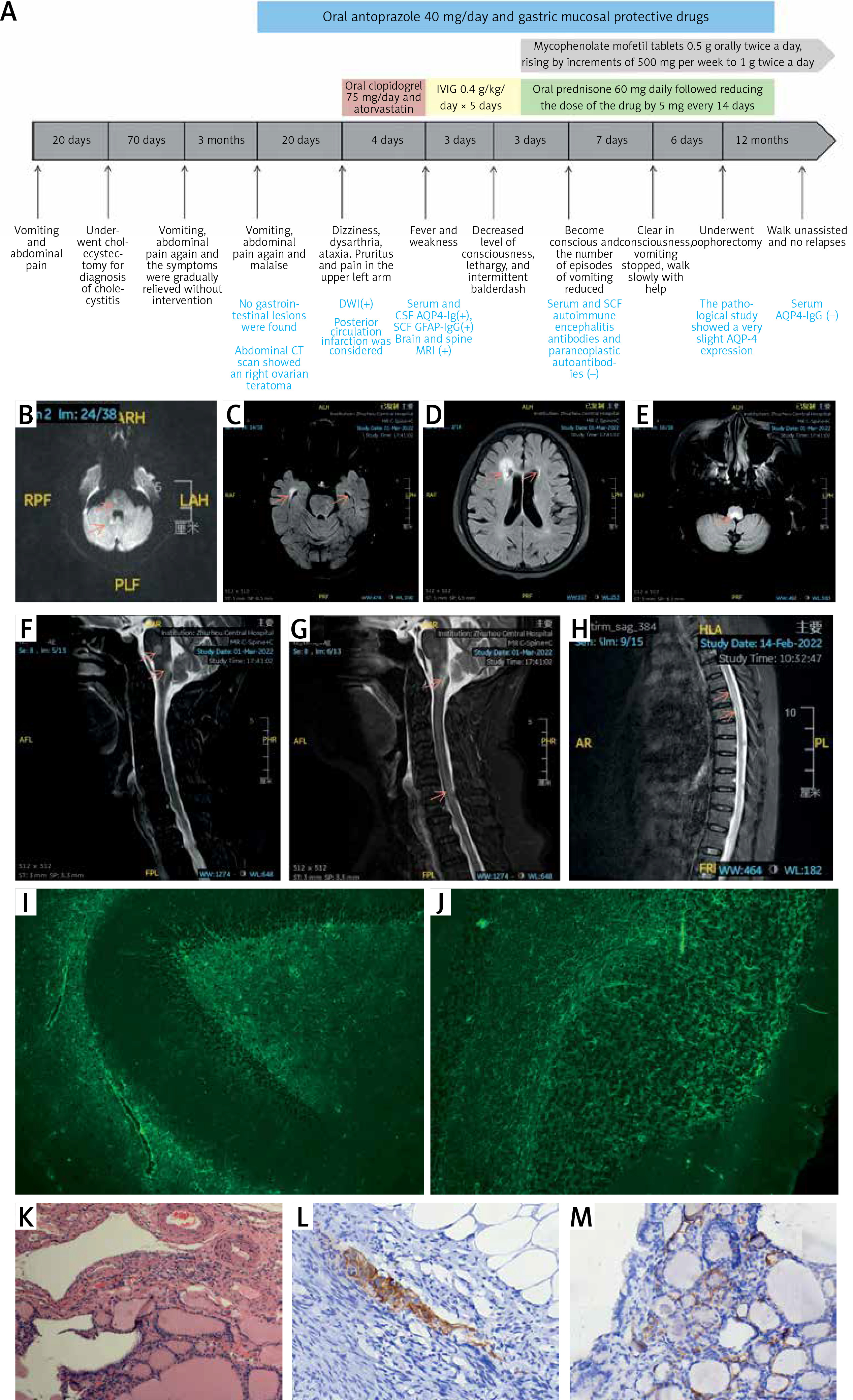
Physical examination revealed that she had dysarthria and horizontal nystagmus sand ataxia with left vision of 20/30 and right vision of 26/30. Her neck was supple, and she felt severe burning pain in the left neck and left upper limb. Pain and tactile sensation relieved below the 6th thoracic vertebra. Her left bicep, triceps, knee, and Achilles reflex were hyperactive with 4-limb strength of 4/5, and the Babinski signs were negative bilaterally.
The patient showed a normal electroencephalogram and an abnormal visually evoked potential with prolonged P100 latency and amplitude. Brain and spine magnetic resonance imaging revealed multiple hyperintense T2-flaire lesions (Figures 1 C–H). The patient experienced severe hypokalaemia-related intractable vomiting without any other routine laboratory investigations revealing. Cell-based immunofluorescence assay (CBA) demonstrated negative serum antibodies against ANA, dsD NA, ANCA, ACA, TgAb, TPO-Ab, MOG, and GFAP, whereas anti-AQP4 antibody was positive with a titre of 1 : 10. Cerebrospinal fluid (CSF) analysis revealed the slight elevation of white blood cells (30/mm3) and protein level (55 mg/dl), normal glucose concentration, and decreased chloride. Anti-AQP4 (1 : 10) was also detected in CSF, whereas oligoclonal band and anti-GFAP antibody with a titre of 1 : 3.2 were only detected in CSF using the CBA method. Representative positive signal of GFAP morphological characteristic, rather than AQP4, from neuroglia was found in the hippocampus and cerebellum, detected using a monkey cerebellar tissue-based assay (TBA) (Figures 1 I, J).
She was finally diagnosed with neuromyelitis optica spectrum disorders (NMOSD), and the symptom of fever developed. The patient received intravenous injection of gamma globulin (IVIG) at the dose of 0.4 g/kg/day for 5 days instead of high-dose glucocorticoid therapy due to lung infection. She rapidly experienced decline in consciousness level, lethargy, and intermittent balderdash at day 3 of IVIG therapy. Thereafter, the patient underwent lumbar puncture again. The results of CBA showed that both serum and CSF autoimmune encephalitis antibodies were negative, as well as paraneoplastic autoantibodies. Furthermore, the patient received oophorectomy, and the pathological study showed a mature cystic teratoma that was AQP-4 negative (Figures 1 I–L). Two days after IVIG treatment under intensive care, the patient became conscious, and the frequency of vomiting reduced. Then, she was treated with prednisone at a dose of 60 mg/day followed by a reduction to 5 mg every 14 days, combined with oral mycophenolate mofetil tablets twice daily at 1 g each. During one-year follow-up, the patient was found to be able to walk independently without further relapses, and the AQP4 in serum turned to negative. The timeline of the patient is shown in Figure 1 A.
NMOSD shows a certain clinical similarity to autoimmune glial fibrillary acid protein astrocytopathy (GFAP-A). Common manifestations of NMOSD include transverse myelitis, optic neuritis (ON), and encephalopathy symptoms if the brain is involved [1]. Transverse myelitis and ON also occur frequently in GFAP-A [2]. Area postrema syndrome (APS) is considered as one of the symptoms of GFAP astrocytopathy [3]. We currently report a rare case of AQP4 and GFAP double-positive NMOSD complicated with ovarian teratoma, presenting with APS, long-segment myelitis, and encephalopathy. Our report aims to increase awareness of the clinical characteristics of GFAP/AQP4-IgG double-positive autoimmune astrocytopathy.
It has been reported that 10% of patients who are GFAP-IgG positive have coexisting AQP4-IgG positivity in serum or CSF [4]. Lin et al. reported a 72-year-old male with overlapping positivity of GFAP-IgG and aquaporin-4-IgG in CSF, who presented with vision loss, hiccups, fever, headache, and ataxia [5]. Unlike this case, our patient showed myelitis without characteristic radial enhancing patterns present. Flanagan et al. described a series of 10 GFAP/AQP4-IgG double-positive cases, 7 of which presented with encephalitis, 2 with encephalomyelitis, and one with neuromyelitis optica [4]. Unfortunately, the research did not disclose any specific clinical characteristics of patients, and no further statistical analysis was performed.
Another research based on Chinese individuals summarized the clinical characteristics of 8 GFAP/AQP4 double-positive cases with myelitis, demonstrating a mean age-at-onset of 37.3 years (range: 23–50 years) [6], much lower than the case we reported (71 years). This may be associated with paraneoplastic factors. The study found that all 8 cases (100%) presented with urination, defecation, and sensory dysfunction. However, the absence of urination and defecation difficulties in our patient is inconsistent with the incidence of 100% stated in the study [6]. In addition, the study demonstrated no one presented with disturbance of consciousness, but the patient we reported had impaired consciousness. These inconsistencies may be contributed to the fact that there was no significant difference in clinical characteristics including headache, disturbance of consciousness, abnormal vision, ataxia, behavioural abnormalities, and MRI showing “radial enhancement” as the specific features of GFAP/AQP4-IgG double-positive patients [6]. Consistent with the features of our patient, there are statistical differences in fever symptom and protein levels in CSF between the GFAP/AQP4-IgG double-positive and AQP4-IgG single-positive groups (p < 0.05) [6]. However, the number of patients included in this study is relatively small; a further study with a large sample size is needed.
There are several case reports of AQP4-IgG-seropositive NMOSD associated with teratomas. Previous research found that teratoma was detected in 7 of 10 patients (70%) with GFAP-IgG and AQP4-IgG double-positive coexisting [4], demonstrating the non-accidental phenomenon of GFAP/AQP4- IgG double-positive accompanied with teratomas. Virgilio et al. reported a 41-year-old woman diagnosed with paraneoplastic neuromyelitis optica, presenting with asthenia, bradycardia (40 bpm), loss of weight, rare vomiting episodes, and AQP4/GFAP double positive in CSF but not in serum [7]. In contrast to the patient we reported, this patient was much younger and had no symptoms related to encephalopathy.
Interestingly, our patient had neither predilection age of NMOSD nor the age characteristics of ovarian teratoma. As suggested by Flanagan et al., teratoma-associated NMOSD probably occurs in younger people [4]. The patient we reported (71 years) was more likely to have paraneoplastic NMOSD associated with other cancers [8]. However, more evidence is required. Considering that diverse neoplasms are usually diagnosed several years after the diagnosis of paraneoplastic NMOSD [9], we will continuously monitor the clinical manifestations of the patient to complement and strengthen our present conclusions. In addition, the patient had a history of COVID-19 vaccination prior to the onset of NMOSD. Because he was an elderly patient with neuromyelitis optica relapsed after receiving 23-valent pneumococcal polysaccharide vaccination [10], the assumption that the vaccine may have been involved in the immunological cascade leading to NMOSD in elderly patients cannot be completely ruled out.
As described by Ikeguchi et al., the clinical features of AQP4-IgG-positive teratoma-associated NMOSD that is GFAP-IgG negative includes the presence of APS, and an increase of IgG in CSF, and the fact that the anti-AQP4 antibody turned negative 6 months after the tumour removal, which are compatible with our current patient [11]. In all 5 reported teratoma-associated NMOSD cases, histopathological analyses of the teratomas revealed the GFAP + neural tissue (glial component) with AQP4 immunoreactivity and infiltration of lymphocytes [4]. However, no AQP4 immunoreactivity in teratoma was observed to support the paraneoplastic aetiology for NMOSD in our current study.
The relationship between teratoma and the development of GFAP/AQP4-IgG double-positive NMOSD remains unclear. HLA-G+ immune cells may be implicated in the complex mechanisms underlying the pathogenesis of inflammatory and autoimmune diseases. HLA-G expression detected in immune cells infiltrating CNS and CSF may contribute to immune regulation in NMOSD [12]. The coexistence of autoimmune disease and tumour may be explained by the immunotolerant role of HLA-G expression in different types of carcinoma, inhibiting the immune responses of MHC-unrestricted NK and MHC restricted T cells [13].
Whether there are specific clinical and histopathological features of AQP4/GFAP-IgG double-positive coexisting with ovarian teratoma is far from clear. Further investigation is required to reveal the potential mechanisms.