Introduction
Worldwide, atherosclerosis (AS) is one of the most common causes of high morbidity and mortality in cardiovascular diseases, with obvious lipid accumulation and chronic inflammation in the aorta [1, 2]. Vascular health and homeostasis depend on the integrity of endothelial cells (ECs) [3]. Endothelial injury occurs in the early stages and contributes to the progression of AS by inducing aggravated inflammatory responses and macrophage recruitment [4]. Moreover, the proliferation of vascular smooth muscle cells (VSMCs) was considered as a crucial factor that led to arterial wall thickening and AS plaque formation [5]. Recent studies indicated that the dysfunction and injury of ECs might result in abnormal phenotypic transition and proliferation which accelerate AS development [6].
Oxidized low-density lipoprotein (ox-LDL) is an important marker of AS [7]. Ox-LDL facilitates the injury and dysfunction of vascular ECs, promoting the pro-adhesion characteristics of ECs and facilitating the recruitment of monocytes. Ox-LDL can promote the formation of atherosclerotic plaques and EC apoptosis [7–9]. Therefore, ox-LDL is a critical factor during the pathogenesis of AS. Moreover, ox-LDL was often used as an in vitro AS cell model inducer to explore the specific genes that participated in AS.
Considered as a subtype of the tropomyosin (TPM) family, TPM2 is an important protein molecule associated with thin filament stabilization in multiple eukaryotes and involved in muscle contraction [10]. There is evidence that TPM2 exerts a role in mediating cellular morphogenesis and proliferation, as well as migration [11]. Scientists found aberrant expression of TPM2 in cancer tissues which regulated tumor progression [12]. Moreover, Kubo et al. indicated that abnormal myosin microfilament homeostasis could damage endothelial cells and promote lipid accumulation [13]. A previous study identified aberrant TPM2 expression in AS plaques [14]. However, reports on the specific regulatory function and mechanism of TPM2 in AS development are lacking.
In this study, we investigated TPM2 expression in the tissues of AS patients and the ox-LDL-treated AS in vitro cell model. An overexpression assay was performed to evaluate the function of TPM2 in ox-LDL-induced AS progression. Furthermore, based on the ras homolog family member A (RhoA) signaling pathway, the regulatory mechanism of TPM2 in AS was explored. Our research could provide a novel theoretical foundation to further understand the pathogenesis of AS and effective therapies.
Material and methods
Patient samples
Pathological and adjacent control samples were collected from 36 AS patients (Yantai Yuhuangding Hospital, April 2018 to October 2019) who had not received any treatment before. Patients with concomitant inflammatory diseases, malignancies and autoimmune diseases or a history of diabetes and thyroid diseases were excluded. Informed consent was obtained from every patient and the research was approved by the ethics committee of Yantai Yuhuangding Hospital, following the Declaration of Helsinki (2008) of the World Medical Association. The tissues were collected during carotid endarterectomy and transported to the laboratory under a low temperature condition (cold saline and ice), and then rapidly dissected into the adjacent control section and atherosclerotic section. The samples were finally stored at –80°C.
Cell culture and treatment
We purchased human aortic endothelial cells (HAECs) and human vascular smooth muscle cells (VSMCs) from ATCC (Manassas, VA, USA). To culture these two kinds of cells, Dulbecco’s modified Eagle’s medium (DMEM, Thermo Fisher Scientific), which was supplemented with 2 mM glutamine, 100 U/ml penicillin and 10% fetal bovine serum (FBS, Gibco BRL, Gaithersburg, USA), was utilized. The atmosphere for cell culture was set at 37°C with 5% CO2. To overexpress TPM2, a pcDNA3.1 plasmid was recruited and the construction of an overexpression vector was performed by Integrated Biotech Solutions (Shanghai, China). The transfection used a pc-TPM2 plasmid (50 nM) for 48 h in line with the protocol of Lipofectamine 3000 (Invitrogen; Shanghai, China). Narciclasine (MedChemExpress, Shanghai, China), an activator of the RhoA pathway, was added to the medium at the concentration of 50 nM.
Human aortic endothelial cells were exposed to Ox-LDL (Yiyuan Biotechnologies, China) at different concentrations (0 μg/ml, 6.25 μg/ml, 12.5 μg/ml, 25 μg/ml, 50 μg/ml, 100 μg/ml, 200 μg/ml) for 24 h or at 100 μg/ml for various periods of time (0 h, 3 h, 6 h, 12 h, 24 h, 48 h, 96 h). The co-culture of HAECs and VSMCs was performed by incubating VSMCs for another 24 h using the conditional medium of HAECs.
MTT assay
After stimulation with ox-LDL, cells were plated into six-well plates and incubated for 24 h. Subsequently, we gently washed the cells three times using phosphate buffer saline (PBS) buffer. MTT solution (5 mg/ml) was supplemented into the culture medium and the cultured cells were incubated in darkness (4 h, 37°C). After that, the cell culture system was dissolved using dimethylsulfoxide (DMSO). Finally, the absorbance at 570 nm of different groups was detected on a Bio-Rad microplate reader (Hercules, USA).
Enzyme-linked immunosorbent assay (ELISA)
After centrifugation, the culture medium was collected and used to detect the contents of interleukin-1β (IL-1β), IL-6 and tumor necrosis factor-α (TNF-α), as well as vascular cell adhesion molecule-1 (VCAM-1), E-selectin, macrophage chemoattractant protein-1 (MCP-1) and C-C motif chemokine ligand 2 (CCL2), in the supernatant according to the method described in the manufacturer’s protocol of human ELISA kits (R&D Systems, Minnesota, USA).
The apoptosis rate of HAECs was evaluated using a Cell Death Detection ELISA kit (Merck, Darmstadt, Germany) which was based on the principle of oligosome generation and the process of DNA fragmentation in apoptotic cells.
Wound healing assay
Once the cells reached 80% confluence, a sterile micropipette tip was used to scratch a wound on the surface of the cell monolayer. Then, fresh medium was supplemented to culture the washed cells. Twenty-four hours later, the migrated cells in the wounded field were measured.
Transwell
Vascular smooth muscle cells after different treatments were seeded onto the upper layer of a Transwell chamber with a diameter of 8 μm (Corning, NY, USA) which was pre-coated with Matrigel. The bottom layer of the chamber was filled with 10% PBS-supplemented DMEM. The incubation lasted for 24 h. Then, the cell invasion ability was determined by counting the number of cells on the surface of the lower layer. The Leica Wild M690 microscope (Leica Microsystems GmbH, Wetzlar, Germany) was utilized for data collection.
Real-time quantitative PCR (RT-qPCR)
After the indicated incubation, the cultured cells were treated using RNAiso Plus (Takara Bio, Dalian, China) to extract total RNA. According to the manufacturers’ instructions of a PrimeScript RT reagent Kit (Invitrogen, Carlsbad, USA), the total RNA was reverse-transcribed into cDNA followed by being rinsed using gDNA Eraser (Takara Bio, Dalian, China). Then, on a platform of the StepOnePlus Real-time PCR System (Applied Biosystems, Foster City, USA), the qPCR procedure was performed in line with the protocol of a Quanti-Nova SYBR Green RT-PCR Kit (Qiagen, Dusseldorf, Germany). GAPDH was set as the internal reference gene in the triplicate repeats. To quantify the final data, the 2–ΔΔCt method was used.
Western blot (WB) analysis
Total protein was obtained with the help of RIPA (radio immunoprecipitation assay) lysis buffer. Protein samples were separated by 10% SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and transferred onto a PVDF (polyvinylidene fluoride) membrane. Members were blocked with 5% skimmed milk mixed in TBST (tris buffered saline tween) and cultured at 4°C overnight with primary antibodies (Abcam, Cambridge, UK), anti-TPM2, anti-CI, anti-CII, anti-CIII, anti-CIV, anti-VCAM-1, anti-E-selectin, anti-MCP-1, anti-CCL-2, anti-PCNA, anti-E-cadherin, anti-MMP-9, anti-GTP-RhoA, anti-RhoA, anti-ROCK1 (Rho-associated coiled-coil containing protein kinase 1) and anti-ROCK2. Then, HRP (horseradish peroxidase)-conjugated secondary anti-body (LI-COR, Lincoln, USA) was applied at room temperature for 1 h. An enhanced chemiluminescence system was employed to detect the protein bands and ImageJ software was used for analysis.
Mitochondrial function and oxidative stress
The mitochondrial membrane potential (MMP, ΔΨ) of HAECs was evaluated according to a previous study [15]. The experiment was based on use of a dual emission potential probe with the JC-1 dyeing method. Briefly, the cultured cells were incubated with JC-1 (10 mg/ml) for 30 min. The absorbances at 527 nm and 590 nm of the incubated cells were recorded under a spectrophotometer (Flex Station384, Molecular Devices, USA).
Reactive oxidative species (ROS), superoxide dismutase (SOD) and lactate dehydrogenase (LDH) were indicated as the cell indexes of oxidative stress. A commercial kit, Reactive Oxygen Species Assay Kit (Beyotime, Shanghai, China) was purchased for ROS detection. The medium-discarded cells were incubated with 10 μM 2′,7′-dichlorofluorescin diacetate for 1 ml (20 min, 37°C, in the dark). The fluorescence intensity was monitored at 488 nm (excitation) and 525 nm (emission). After being lysed and centrifuged, the supernatant for cell culture was supplemented with SOD working fluid and incubated for 15 min. Then, the absorbance at 520 nm was detected on a microplate reader using a SOD Activity Detection kit (Solarbio, Beijing, China). To evaluate LDH content in the medium supernatant, the cells (1 × 104 cells/ml) were transferred to fresh 96-well plates and pretreated using LDH buffer for 2 h. Then, the detection procedure was performed and data were counted at 490 nm according to the instructions of the ABSBio LDH Activity Detection kit (CNhaoBio, Beijing, China).
Statistical analysis
All data were analyzed with GraphPad Prism 8 and represented as the mean ± standard deviation (SD) obtained from at least three repeats. Statistical comparisons were completed by one-way analysis or Student’s t-test (unpaired). P < 0.05 was considered to indicate statistical significance.
Results
Tropomyosin 2 is downregulated during AS progression
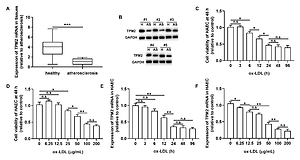
To investigate the potential function of TPM2 in AS development, we first evaluated its expression profile in AS tissues and an in vitro cell model. As shown in Figure 1 A, the mRNA level of TPM2 in AS samples was dramatically decreased compared with healthy ones. Also, TPM2 protein expression was downregulated in 5 randomly selected AS specimens compared with the paired healthy samples (Figure 1 B). After ox-LDL stimulation, the viability of HAECs was inhibited in a dose- and time-dependent manner (Figures 1 C and D). Simultaneously, TPM2 was also downregulated in the ox-LDL-induced AS cell model (Figures 1 E and F).
Figure 1
TPM2 expression in AS tissues and ox-LDL-treated HAEC. A – The mRNA expression levels of TPM2 in healthy tissue and atherosclerosis (AS) samples were evaluated using RT-qPCR. B – Western blot assay showed TPM2 protein expression in healthy and AS tissues. HAEC cells were incubated with ox-LDL treatment at various concentrations (0 μg/ml, 6.25 μg/ml, 12.5 μg/ml, 25 μg/ml, 50 μg/ml, 100 μg/ml, 200 μg/ml) for different times (0 h, 3 h, 6 h, 12 h, 24 h, 48 h, 96 h). C and D – Cell viability was measured using the MTT method. E and F – Expression of TPM2 in ox-LDL-treated HAEC was assessed using RT-qPCR. *p < 0.05, **p < 0.01, ***p < 0.005; n.s. – not significant
Tropomyosin 2 overexpression inhibited inflammation and pro-adhesion responses in ox-LDL-treated HAECs
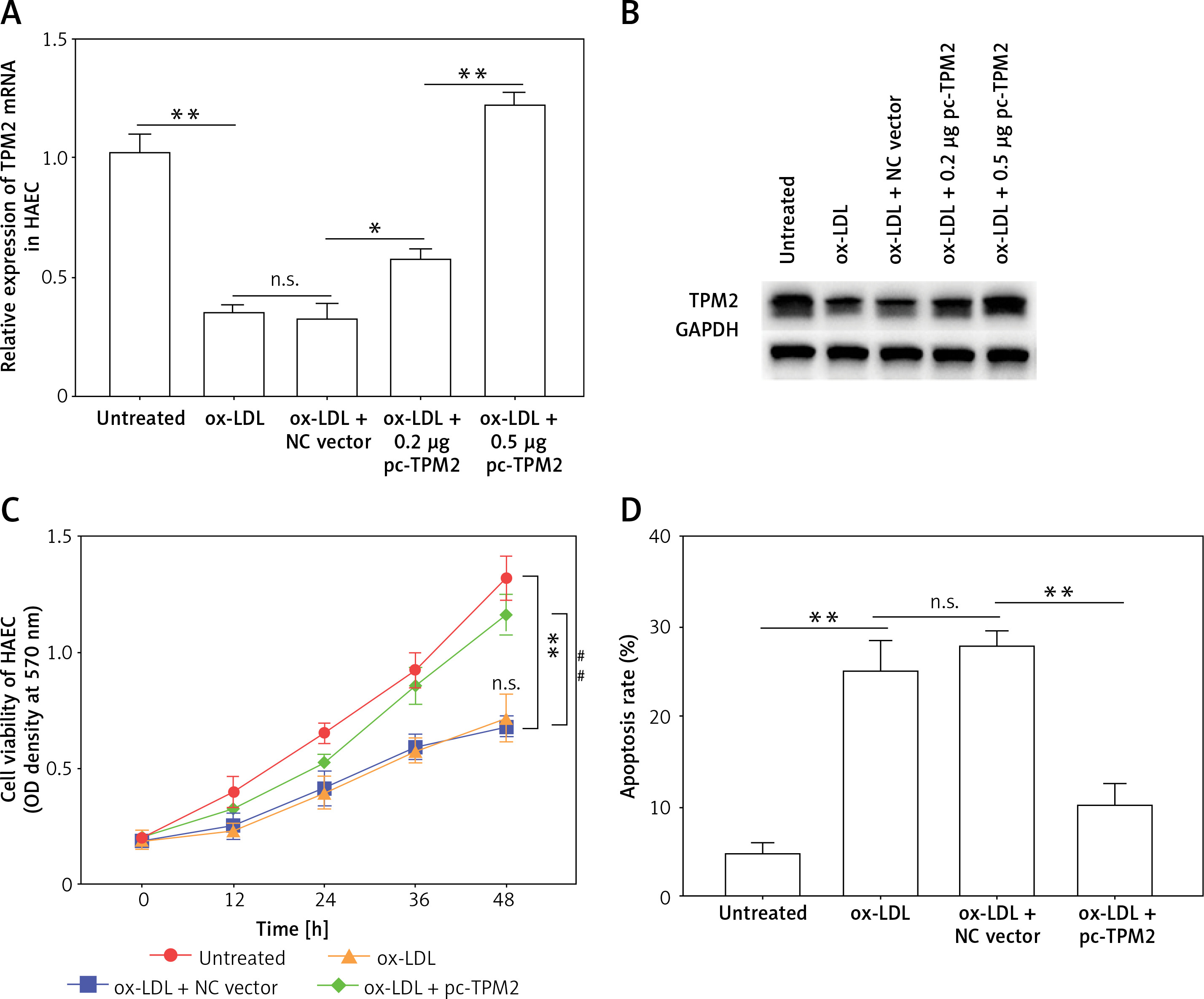
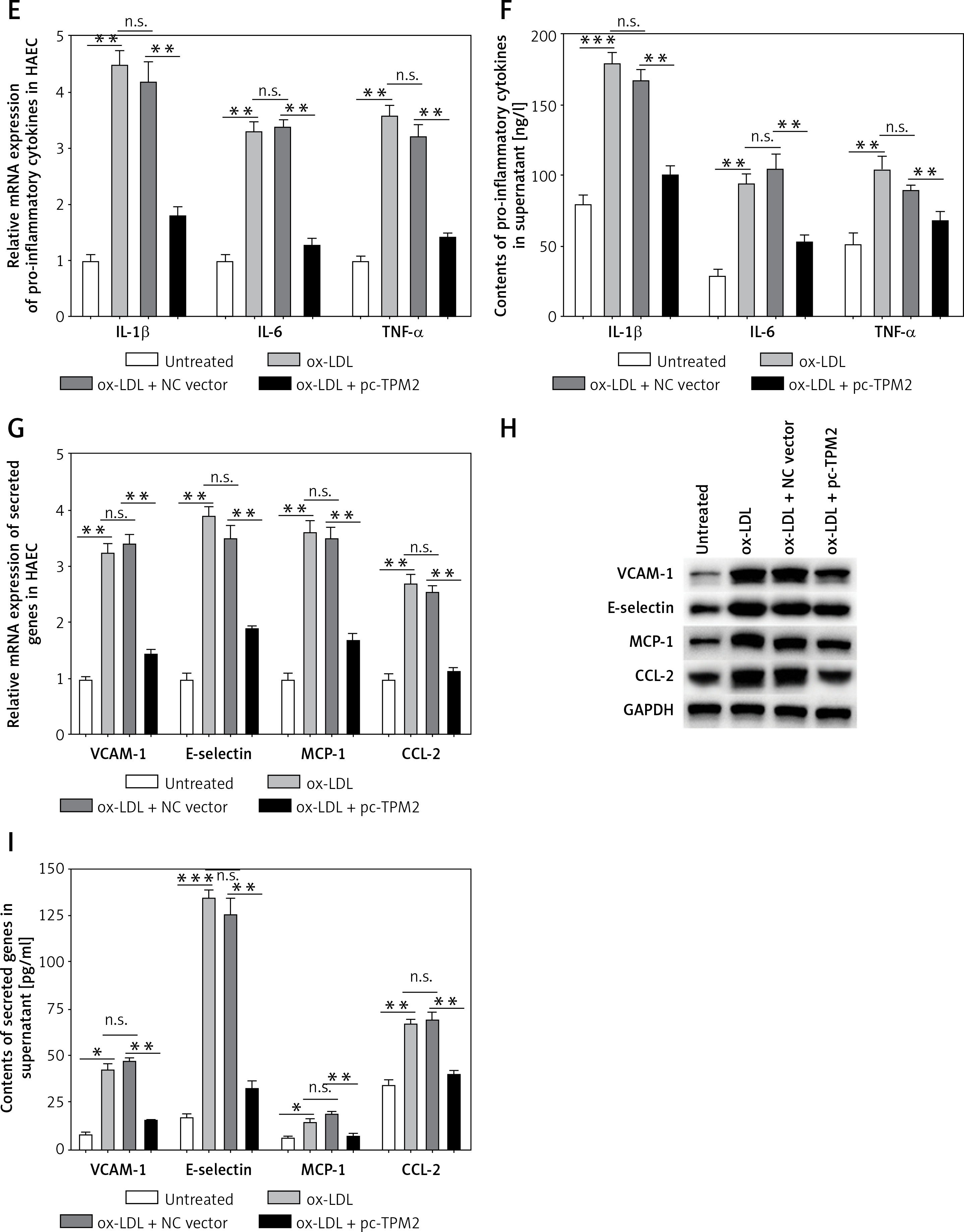
Subsequently, an overexpression assay was performed by using the constructed TPM2 plasmid. The results showed that transfection of the pc-TPM2 vector significantly promoted TPM2 expression in HAECs under ox-LDL treatment (Figures 2 A and B). We also found depressed viability and elevated apoptosis of HAECs after ox-LDL stimulation, which were reversed by TPM2 overexpression (Figures 2 C and D). Studies have demonstrated that the inflammation and adhesion responses, as well as monocyte chemotaxis, of vascular endothelial cells had important roles in AS progression [6]. As indicated in Figures 2 E and G, the expression levels of inflammatory (IL-1β, IL-6, TNF-α), adhesive (VCAM-1, E-selectin) and chemokine genes (MCP-1, CCL-2) were dramatically upregulated in the ox-LDL-induced AS in vitro model, and inhibited by TPM2 overexpression. The contents of IL-1β, IL-6 and TNF-α in the supernatant of the ox-LDL group were obviously higher than those in the control group, while the indexes were lower in the group of ox-LDL + pc-TPM2 compared with the ox-LDL group (Figure 2 F). In addition, the elevated protein expression and production of adhesion molecules and chemokines, induced by ox-LDL, were also decreased by TPM2 overexpression (Figures 2 H and I).
Figure 2
Effects of TPM2 overexpression on inflammation and adhesion responses of HAEC. HAEC cells were treated with ox-LDL (100 μg/ml) for 24 h followed by transfections of NC vector and pc-TPM2 (0.2 μg and 0.5 μg; 48 h). A and B – Transfection efficiency of pc-TPM2 was evaluated using RT-qPCR and western blot. C – MTT assay was used to evaluate cell proliferation.**p < 0.01 vs. untreated group (n.s. – not significant) vs. ox-LDL group; ##p < 0.01 vs. ox-LDL + NC vector group. D – Cell apoptosis rate was measured using a cell death ELISA kit. E and G – RT-qPCR was performed to analyze mRNA expression of inflammatory and adhesive factors. F and I – Contents of cytokines and adhesion molecules in supernatant were measured using ELISA kits. H – Western blotting showed protein expression. *p < 0.05, **p < 0.01, ***p < 0.005, n.s. – not significant
Tropomyosin 2 overexpression inhibited oxidative stress and mitochondrial dysfunction in ox-LDL-treated HAECs
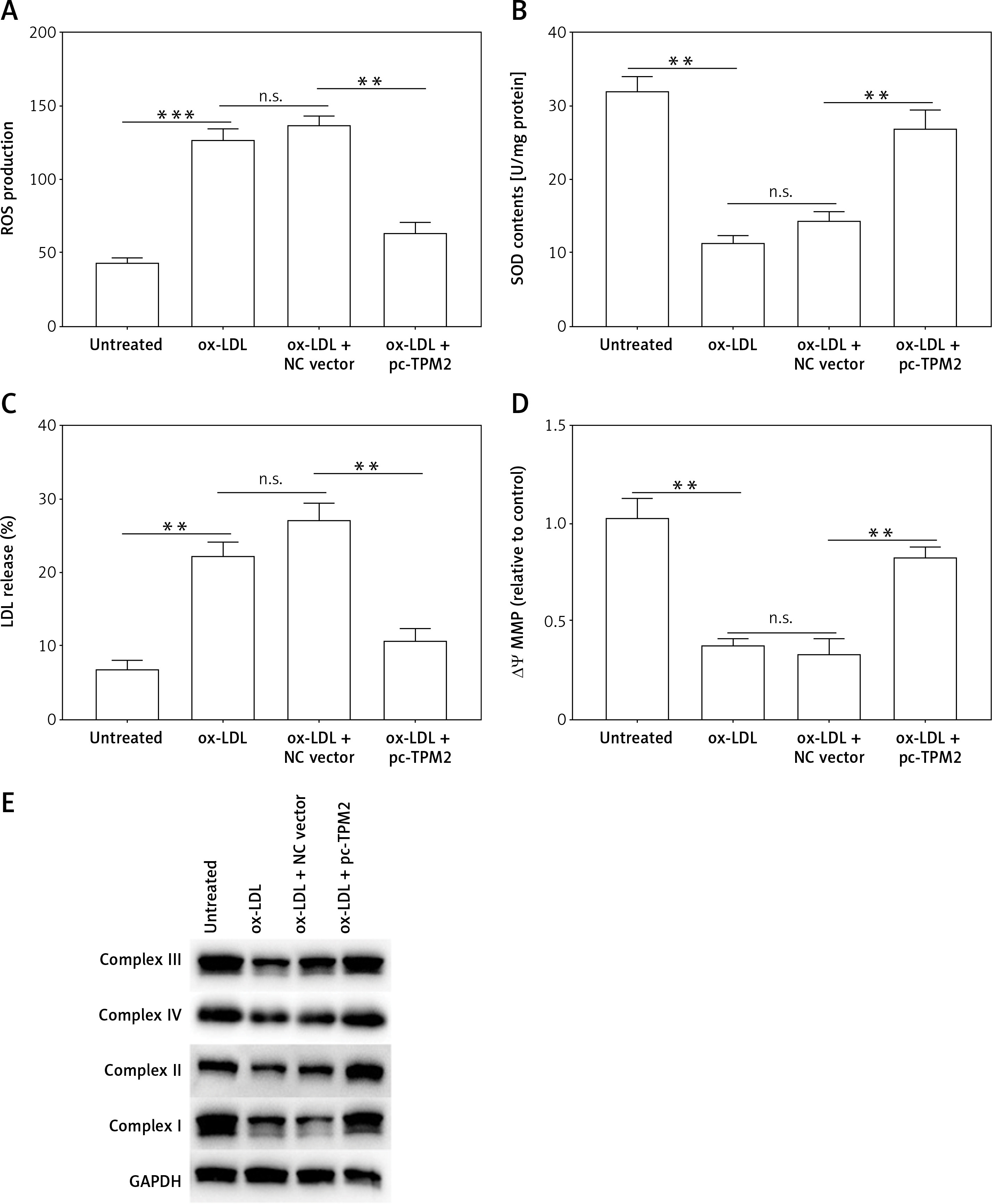
Oxidative stress and mitochondrial dysfunction are crucial factors that participate in the mediation of endothelial cell function and AS progression [16]. The next experiments evaluated the oxidative stress indexes and mitochondrial function of cells under different treatments. First, ROS production and LDH release, the key features of oxidative stress in cells, were both promoted by ox-LDL, while SOD content was decreased; these unfavorable effects were depressed by TPM2 overexpression (Figures 3 A–C). Additionally, the MMP of mitochondria in ox-LDL-induced HAECs was dramatically decreased and respiratory complex protein expression was suppressed; overexpressing TPM2 also reversed the undesired impacts of ox-LDL (Figures 3 D and E).
Figure 3
Effects of TPM2 overexpression on oxidative stress and mitochondrial dysfunction of HAEC. pc-TPM2 plasmids were transfected into HAEC cells (0.5 μg; 48 h) after ox-LDL treatment (100 μg/ml; 24 h). A–C – Commercial detection kits were used to determine the contents of ROS, SOD and LDH secreted by HAEC under different treatments. D – MMP was detected using the method based on the JC‑1 dyeing dual emission potential probe. E – Western blotting showed the expression of mitochondrial respiratory complex proteins. **p < 0.01, ***p < 0.005, n.s. – not significant
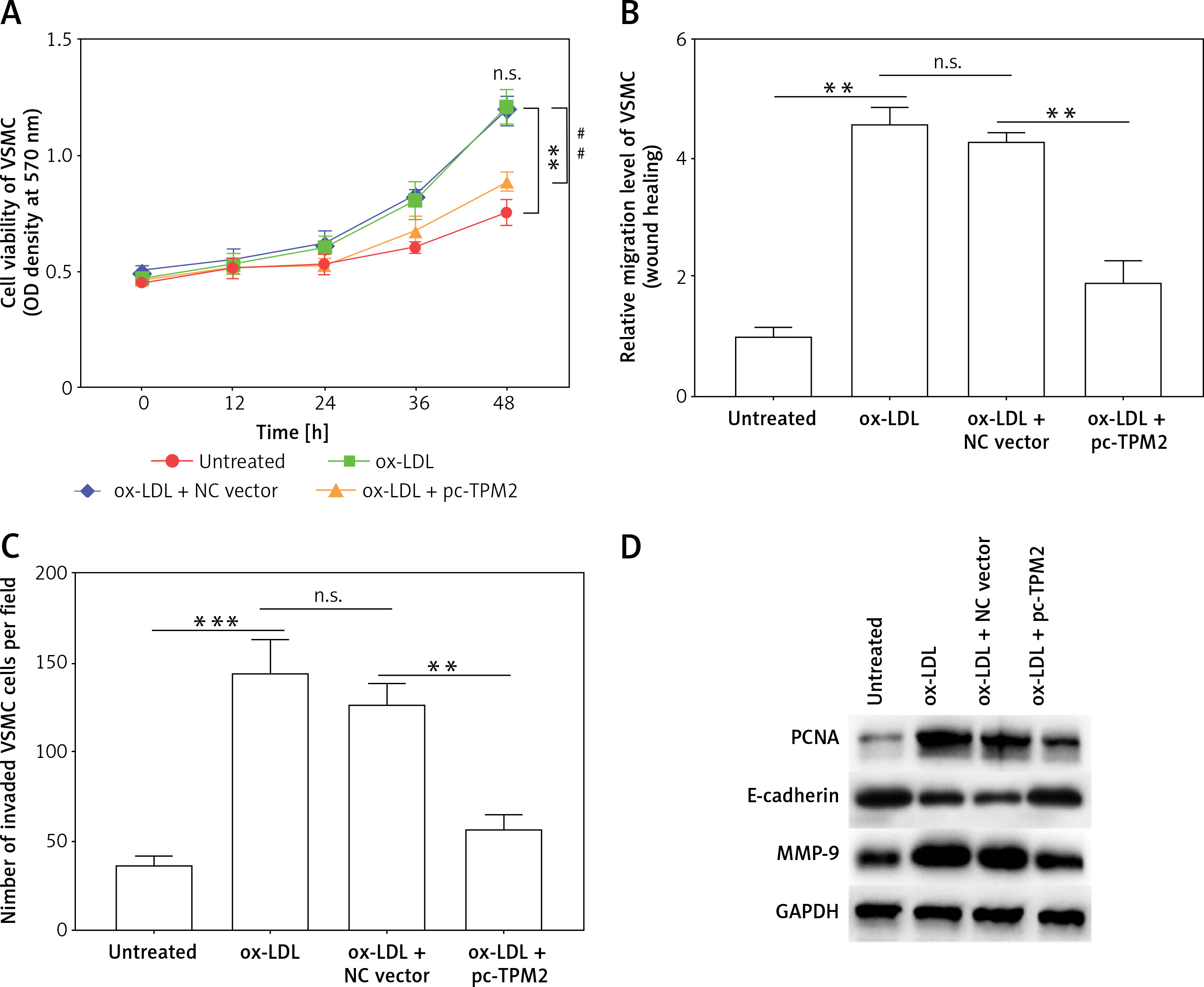
Tropomyosin 2 regulated proliferation, migration, and invasion of VSMCs affected by injured HAECs
The over-activation of VSMCs was an important contributor to AS development. As shown by the results, the cell proliferation, migration and invasion of VSMCs in ox-LDL group were significantly promoted compared with the control group; and TPM2 overexpression depressed these effects of ox-LDL (Figures 4 A–C). Moreover, the expression of proliferation, migration and invasion marker genes (proliferating cell nuclear antigen [PCNA], E-cadherin, matrix metallopeptidase 9 [MMP-9]) was upregulated, consistent with the induced cell processes in the ox-LDL-induced AS VSMC cell model and suppressed by TPM2 overexpression (Figure 4 D).
Figure 4
TPM2 plays a role in the growth and migration of ox-LDL-treated HAECs via the RhoA pathway. HAECs were induced by 100 μg/ml ox-LDL for 24 h. The culture media of HAEC under various treatments were isolated and subsequently used to culture VSMCs for 24 h. Then, the next indexes of VSMCs were measured. A – MTT assay analyzed cell proliferation. **p < 0.01 vs. untreated group; n.s. (not significant) versus ox-LDL group; ##p < 0.01 vs. ox-LDL + NC vector group. B and C – Cell migration and invasion were evaluated using wound healing and Transwell methods. D – Western blotting was performed for the detection of protein expression. **p < 0.01, ***p < 0.005, n.s. – not significant
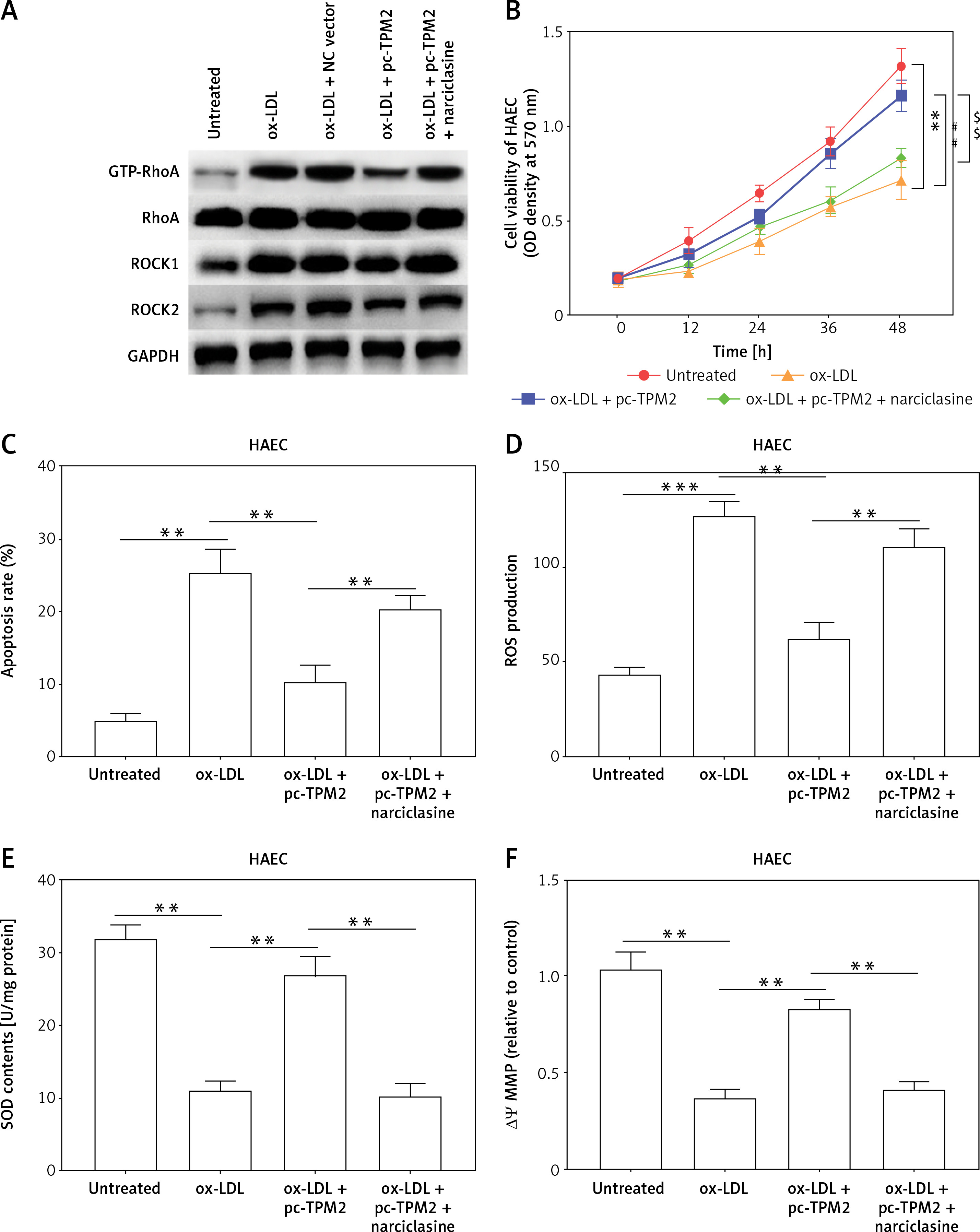
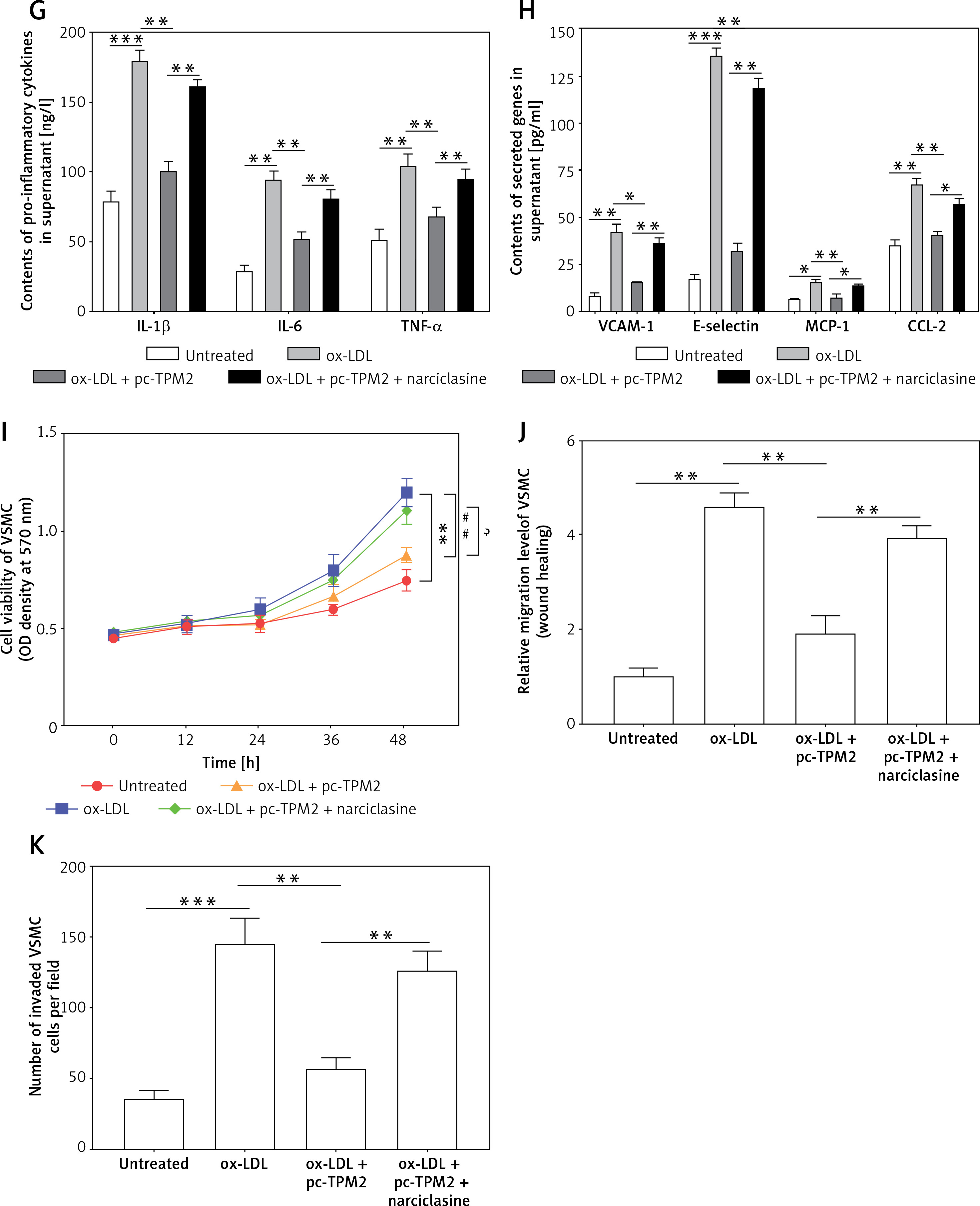
Tropomyosin 2 mediated ox-LDL-induced AS progression via RhoA signaling in vitro
Previous studies indicated that RhoA signaling played an important role in the development of AS [17]. As our results show in Figure 5 A, the expression levels of key proteins in RhoA signaling, GTP-RhoA, ROCK1 and ROCK2 were dramatically elevated by ox-LDL, while TPM2 depressed these effects. Using an agonist, narciclasine, we evaluated the function of RhoA signaling in TPM2-mediated AS progression. The improved cell viability, SOD content and mitochondrial function, induced by TPM2 overexpression in ox-LDL-treated HAECs, were depressed again when treated with narciclasine, while the inhibited apoptosis and ROS production were elevated (Figures 5 B–F). The production of cytokines, adhesion molecules and chemokines in ox-LDL-treated HAECs of the pc-TPM2 and narciclasine group was significantly higher than that in the TPM2-overexpressed group (Figures 5 G and H). Also, re-activating RhoA signaling hindered the inhibitory effects of TPM2 on ox-LDL-induced abnormal cell proliferation, migration and invasion of VSMCs (Figures 5 I–K).
Figure 5
Impact of RhoA signaling on TPM2-mediated AS progression. HAEC cells were transfected with pc-TPM2 along with narciclasine before being treated with ox-LDL. The isolated medium was subsequently used to culture VSMCs for 24 h. A – Western blotting presented the protein expression of RhoA genes. B and I – MTT was used to determine cell proliferation. **p < 0.01 vs. untreated group; ##p < 0.01 vs. ox-LDL group; $$p < 0.01 vs. ox-LDL + pc-TPM2 group. C, G and H – ELISA kits were utilized for cell death and cytokine content detection. D and E – ROS and SOD contents were measured using commercial kits. F, J and K – JC-1 dyeing, wound healing and Transwell assays were performed to evaluate MMP and cell migration, as well as invasion. *p < 0.05, **p < 0.01, ***p < 0.005, n.s. (not significant). B and I – MTT was used to determine cell proliferation. **p < 0.01 vs. untreated group; ##p < 0.01 vs. ox-LDL group; $$p < 0.01 vs. ox-LDL + pc-TPM2 group. C, G and H – ELISA kits were utilized for cell death and cytokine content detection. D and E – ROS and SOD contents were measured using commercial kits. F, J and K – JC-1 dyeing, wound healing and Transwell assays were performed to evaluate MMP and cell migration, as well as invasion. *p < 0.05, **p < 0.01, ***p < 0.005, n.s. – not significant
Discussion
As one of the most common inflammatory diseases affecting people’s health at present, AS is a root cause of multiple cardiovascular problems, such as stroke, myocardial infarction and gangrene [18]. Increasing evidence suggests that vascular homeostasis would be impacted by the abnormal function of endothelial cells and the dysfunction of vascular smooth muscle cells [3, 19]. During the progression of AS, the aberrant accumulation of ox-LDL disturbed vascular endothelial cell function, including abnormal cell apoptosis and viability, elevated cell oxidative stress, and the increased secretion of various adhesion cytokines, such as intercellular cell adhesion molecule-1 (ICAM-1), VCAM-1 and E-selectin, which led to enhanced infiltration of inflammatory cells [20, 21]. Therefore, attenuating endothelial injury has been considered as an effective way to alleviate AS. Geng et al. reported that the protection of rosuvastatin against AS was achieved by attenuating ox-LDL-induced apoptosis and oxidative stress in endothelial cells [22]. Adropin, a peptide hormone, effectively inhibited ox-LDL-stimulated endothelial cell inflammation and adhesion of monocytes to endothelial cells, further mediating AS progression [6]. Moreover, a recent study demonstrated that mitochondrial abnormality played a crucial role in endothelial cell function during the progression of AS [23]. Song et al. reported that the therapeutic efficiency of gypenoside in treatment of AS was achieved by modulating endothelial apoptosis and affecting mitochondria function in vivo [16]. In our study, the oxidative stress, cell apoptosis, immune cell adhesion and mitochondrial dysfunction in ox-LDL-induced AS endothelial cells were significantly depressed by promoting TPM2, which was consistent with previous findings.
Interestingly, we also found that TPM2 inhibited the hyper-activated cell proliferation, migration and invasion of VSMCs which were induced by ox-LDL-stimulated endothelial cells. Under the stimulation of injured endothelial cells in AS, VSMCs presented aberrant hyperplasia accelerating AS progression [24]. The promoted effects of miR-93 on cardiovascular disease, including AS, were achieved by enhancing cell proliferation and migration of VSMCs [25]. A compelling study demonstrated that, by mediating mitophagy, apelin-13 elevated the proliferation and migration abilities of VSMCs, which exacerbated the lesion and development of AS [26]. Moreover, in the research of Lin et al., Notch3 was detected in the exosomes of high glucose-induced endothelial cells and exosomes containing Notch3 were taken up by VSMCs to mediate the calcification and aging of VSMCs [27]. Forkhead box protein M1 (FoxM1) in VSMCs was activated by factors secreted from dystrophic endothelial cells and led to disadvantageous cell proliferation, which contributed to vascular remodeling and hypertension in the pulmonary artery [28]. Zheng et al. reported that VSMCs produced miR-155, packaged in exosomes, which showed harmful effects on the integrity of endothelial cells, accelerating plaque formation in blood vessels and atherogenesis [29]. According to the above evidence, we inferred that the interaction between endothelial cells and VSMCs during AS progression might occur in a paracrine manner. However, further investigations should be performed to illustrate how, and in which specific manner, TPM2 participates in the regulation of endothelial cells affecting the function of VSMCs under AS conditions.
Previously, TPM2 emerged as a crucial suppressor in various tumor diseases. In a study based on colorectal cancer, decreased expression of TPM2 was verified in patient tissues and was considered as a valuable target for the early diagnosis of colorectal cancer [30]. Downregulated TPM2 was found in breast cancer cells and the low expression of TPM2 could independently predict the outcome and survival of cancer patients; moreover, silencing TPM2 promoted the resistance of tumor cells to paclitaxel, an anti-tumor drug [31]. However, studies have demonstrated that the TPM2 gene encodes β-tropomyosin, a member of the actin filament binding protein family, which is mainly expressed in slow, type 1 muscle fibers [32]. Diseases associated with TPM2 also include nemaline myopathy 4 and arthrogryposis, distal, type 1A, as well as dilated cardiomyopathy and cardiac muscle contraction [33, 34]. A previous study indicated that TPM2-mediated myosin microfilament homeostasis participated in the regulations of endothelial cell function and lipid accumulation [13]. Recently, it was suggested that high-temperature superconducting, induced by TPM2, might depress the formation of foam cells and minimize the accumulation of lipoproteins in AS [14, 35, 36]. Meng et al. investigated two datasets focused on AS and identified the dramatical abnormity of TPM2 among differentially expressed genes, regarding TPM2 as a potential biomarker for AS diagnosis and therapy [14]. Our study further explored the specific role and regulatory mechanism of TPM2 during AS development to validate and strengthen the evidence that TPM2 was used as a potential predictive biomarker for AS prognosis or therapy which might be of guiding significance for clinical research.
Additionally, the present study is the first to discover that RhoA signaling is a crucial bridge through which TPM2 mediates AS progression. Previously, Cui et al. demonstrated that silenced TPM2 aberrantly enhanced RhoA activation, and then aggravated tumor growth in colorectal cancer [37]. However, RhoA signaling was proved to be hyper-activated and involved in various cardiovascular diseases. For example, regulating RhoA/ROCK signaling was an effective way to impact the function of blood vessel and other vascular problems [38]. Further evidence suggested that sustained activation of the RhoA pathway was induced by miR-3188 downregulation and promoted ox-LDL-induced AS development [39]. Targeting the ROR2/RhoA pathway was a promising method for AS therapy due to its effect on promoting the cell viability and motricity of VSMCs [17]. Consistently, in this research, the RhoA/ROCK pathway was also depressed by TPM2, which led to the attenuation of ox-LDL-induced endothelial injury and dysfunction of VSMCs, finally alleviating AS progression.
However, limitations also restricted our research. For example, more investigations are still needed to further explore the mechanism of how TPM2-induced HAEC dysfunction affects the action of VSMC cells during AS progression. The factors causing TPM2 abnormality in the pathological process of AS are still unclear. To strengthen the evidence that TPM2 plays a key role in AS development, it should be verified in vivo using a constructed rat model.
In conclusion, we identified abnormal TPM2 downregulation in both AS tissues and the ox-LDL-induced cell model. Additionally, overexpressing TPM2 depressed the development of AS by regulating cell apoptosis, mitochondrial dysfunction, oxidative stress and inflammatory/adhesive responses of vascular endothelial cells, as well as mediating the activity of VSMCs. More importantly, RhoA signaling was involved in TPM2-regulated AS progression. Generally, a new target for AS therapy was provided according to our evidence.