Familial amyloidosis of Finnish type (FAF), also known as hereditary gelsolin amyloidosis (HGA), is an autosomal dominantly inherited disease caused by mutations in the gelsolin (GSN) gene located at chromosome 9q33.2 [1]. The symptoms of FAF are multifaceted, and the most common symptoms include cranial neuropathy, corneal lattice dystrophy, distal sensorimotor neuropathy, and skin changes [2]. Rare cases presenting with proteinuria, nephrotic syndrome, and end-stage renal failure have been reported [3]. According to the HGMD database, there have been only about thirty reported cases of FAF caused by GSN gene mutations, and thrombotic microangiopathy has not been previously reported in FAF patients. Here we report the case of Finnish renal amyloidosis with renal involvement as the first manifestation, in which the patient also developed thrombotic microangiopathy. Combining with the literature, we discuss the clinical and pathological features and diagnosis of this disease, with a view to deepening the understanding of this type of amyloidosis.
An 11-year-old Chinese girl presented with vomiting and petechiae on both lower limbs for 4 days, followed by a single episode of epileptic seizure. She was admitted to the Hainan Affiliated Hospital of Hainan Medical University on February 12, 2024. Before admission, she had been treated at a local hospital, where routine blood tests revealed hemoglobin of 61 g/l (↓) and platelets of 5 × 109/l (↓). She received methylprednisolone 800 mg/day for 3 days and intravenous immunoglobulin (IVIG) 2 g/kg for 3 days, but showed no significant improvement.
Past medical and family history: The patient was hospitalized in 2017 for edema. At that time, laboratory findings revealed albumin 14 g/l (↓), urine protein 3+ (24-hour urine protein 1.417 g/l) (↑), cholesterol 13.7 mmol/l (↑), and creatinine 62 µmol/l (→). She was diagnosed with nephrotic syndrome, and a kidney biopsy revealed focal segmental glomerulosclerosis-like lesions (Figure 1). The patient was treated with prednisone and tacrolimus for 3 years, leading to symptom improvement, after which treatment was discontinued. Her parents were non-consanguineous and phenotypically normal.
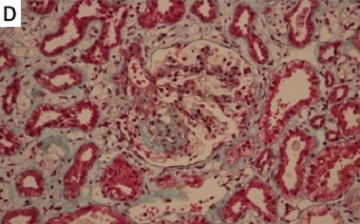
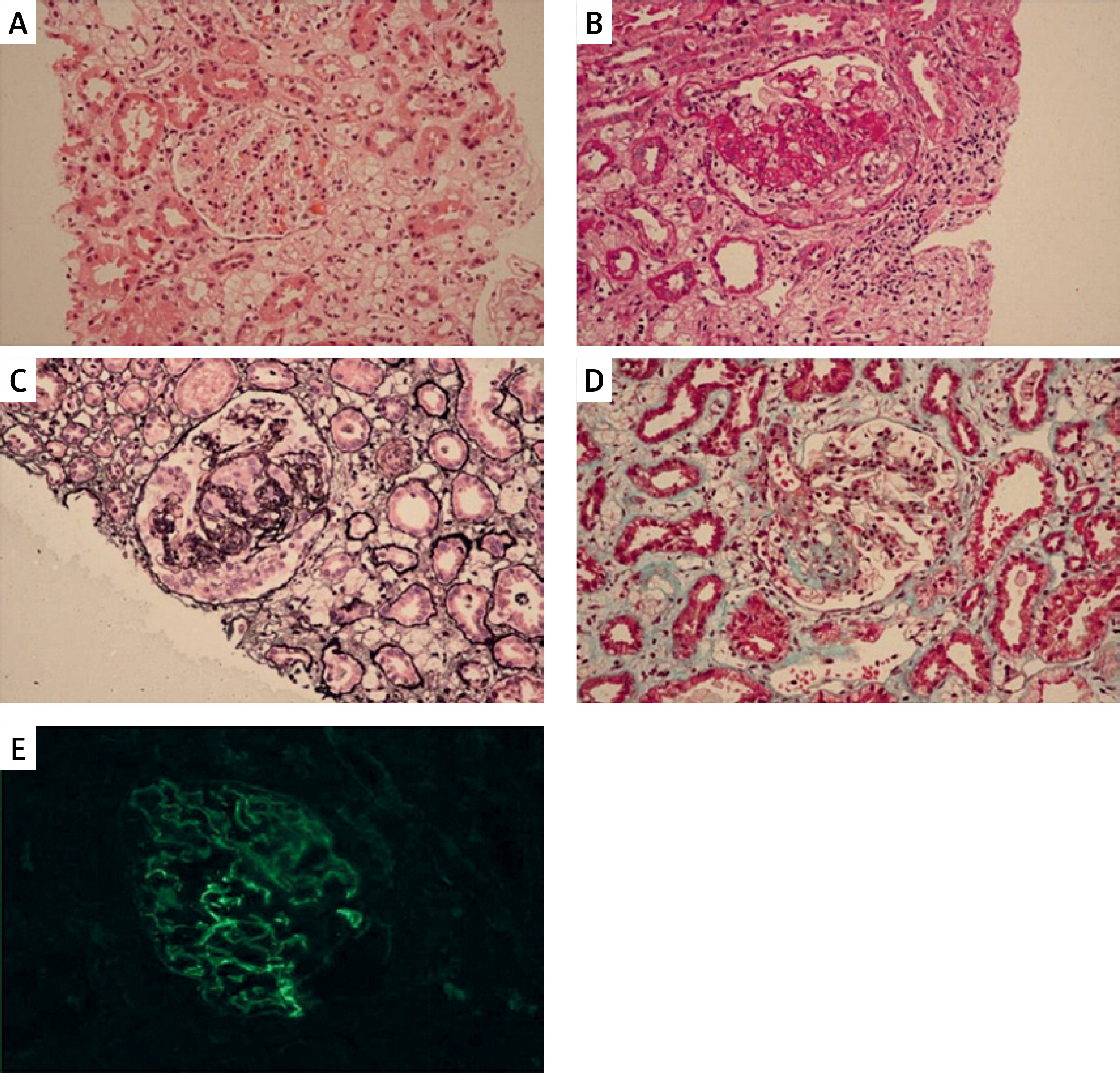
Figure 1
Renal biopsy findings. A – H&E staining showing segmental sclerosis (arrow). B – PAS staining demonstrating mesangial expansion. C – PASM highlighting thickened glomerular basement membranes. D – Masson’s trichrome revealing interstitial fibrosis. E – Negative immunofluorescence for IgG. Scale bars = 50 µm
Height 129.0 cm, weight 27.5 kg, blood pressure 107/81 mm Hg, heart rate 83 beats/min, respiratory rate 20 breaths/min, body temperature 36.6°C. Physical examination revealed an anemic appearance with pale conjunctiva and skin. Multiple petechiae and purpura were observed on both lower limbs, particularly concentrated around the ankles and shins. She had no cardiac, ophthalmic, neurologic, or skin abnormalities on physical exam.
The complete blood count showed hemoglobin 66 g/l (↓) and platelets 13 × 109/l (↓). Urinalysis showed an elevated red blood cell count (4000/l ↑). Stool routine was normal. Coagulation profile indicated a prolonged thrombin time (27.4 s ↑), decreased fibrinogen levels (1.44 g/l ↓), and elevated D-dimer (1.29 µg/ml ↑). Biochemical analysis demonstrated elevated total bilirubin (54.04 µmol/l ↑), with direct bilirubin at 12.91 µmol/l (↑) and indirect bilirubin at 41.13 µmol/l (↑). Other notable findings included significantly elevated lactate dehydrogenase (1949.3 U/l ↑), α-hydroxybutyrate dehydrogenase (1401.8 U/l ↑), and creatine kinase-MB (39.5 U/l ↑), while urea levels were normal (7.81 µmol/l). Cerebrospinal fluid analysis revealed normal routine and biochemical parameters, with a negative etiological test.
Additional laboratory tests showed a normal erythrocyte sedimentation rate, negative direct anti-human globulin test, serum acidified hemolysis test, ENA polypeptide, lymphocyte subsets, and immunoglobulin five-panel. Complement levels and plasma ADAMTS13 activity were normal, with no detected plasma ADAMTS13 inhibitor or IgG. Viral serologies, including influenza A/B, hepatitis B and C, HIV, Epstein-Barr virus, syphilis, rubella, cytomegalovirus, Toxoplasma gondii, and herpes simplex virus, were all negative. Thyroid function tests were normal. Bone marrow examination findings were unremarkable.
Imaging studies: Abdominal ultrasound showed normal liver, bile ducts, spleen, pancreas, and urinary system. Thyroid and gynecological ultrasounds were normal. Both ECG and chest CT results were normal. Renal ultrasound was not performed for this patient. Brain MRI revealed multiple abnormal signals under the bilateral temporal cortex, consistent with acute cerebral infarction (Figure 2).
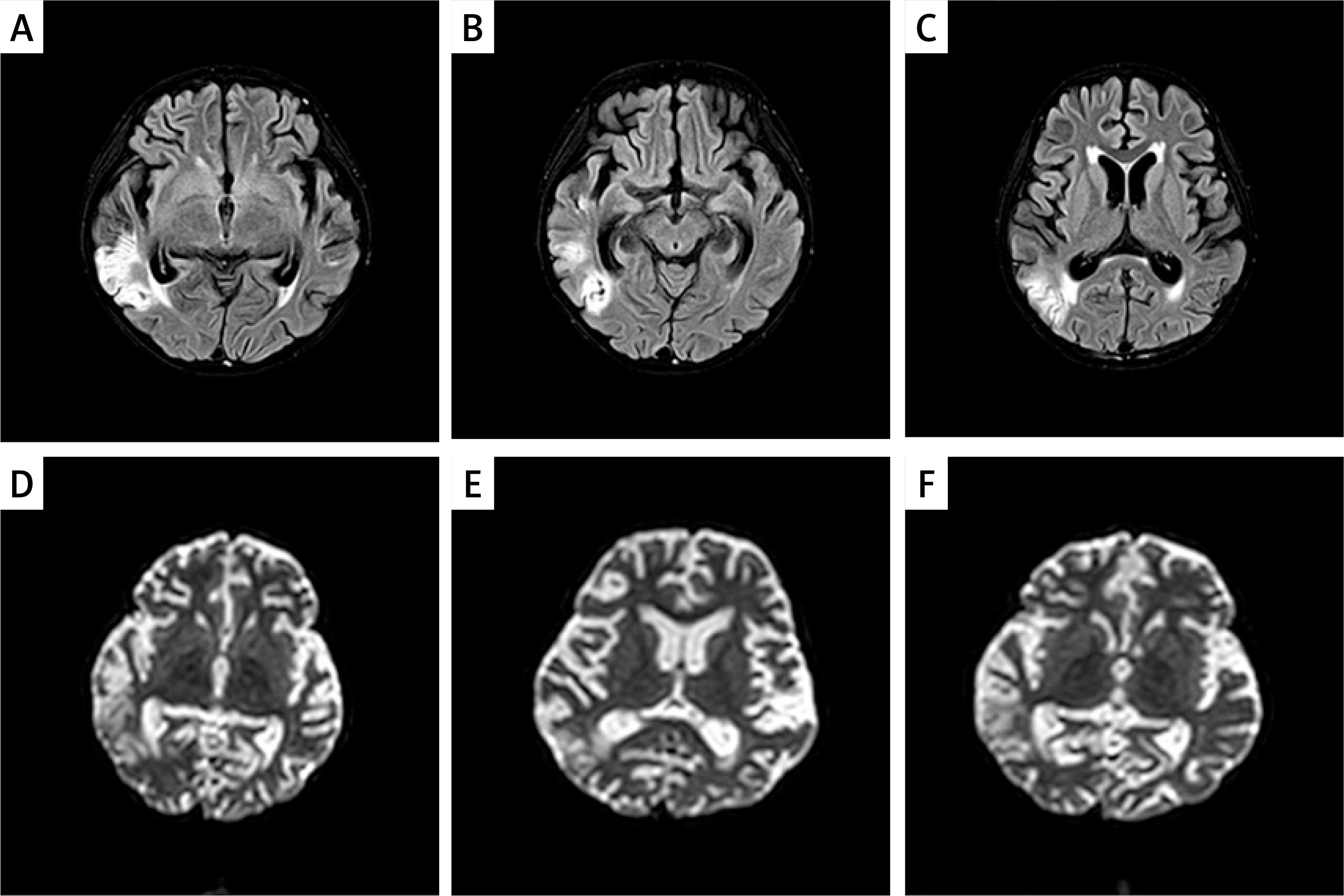
Figure 2
The MRI images of the proband’s brain are shown. A–C – Axial T2-weighted images showing hyperintense lesions in bilateral temporal lobes. D–F – Diffusion-weighted imaging (DWI) demonstrating restricted diffusion in the same areas, consistent with acute cerebral infarction
Whole-exome sequencing (WES): Peripheral blood was collected using EDTA anticoagulant tubes, and DNA was extracted with the TianGen DNA extraction kit. After measuring DNA concentration (Thermo Fisher Scientific, USA), library construction was performed, followed by target capture using the IGT xGen Exome Research Panel v1.0 (Illumina NovaSeq Xplus, USA). Sequencing data in FASTQ format were analyzed using GRCh37/hg19 as the reference genome. The data were filtered with fastp (version: 0.21.0), aligned with bwa (version: 0.7.17), and corrected with dbsnp159. Sequencing depth and coverage were assessed with samtools depth and GATK Depth Of Coverage, ensuring ≥ 100X depth and ≥ 95% coverage above 20X. Variant calling was done with GATK 3.8, and GC content was calculated using mosdepth 0.2.5. Point mutations were annotated with ANNOVAR and local databases, including HGMD and HPO. Specific gene mutations were validated by Sanger sequencing, with primers designed using Primer-BLAST. After amplification, electrophoresis and gel purification were performed, followed by bidirectional sequencing using an ABI platform (Applied Biosystems, USA). The results were analyzed with Chromas software, and the pathogenicity of the mutation sites was assessed based on the patient’s phenotype, genetic features, and other related factors.
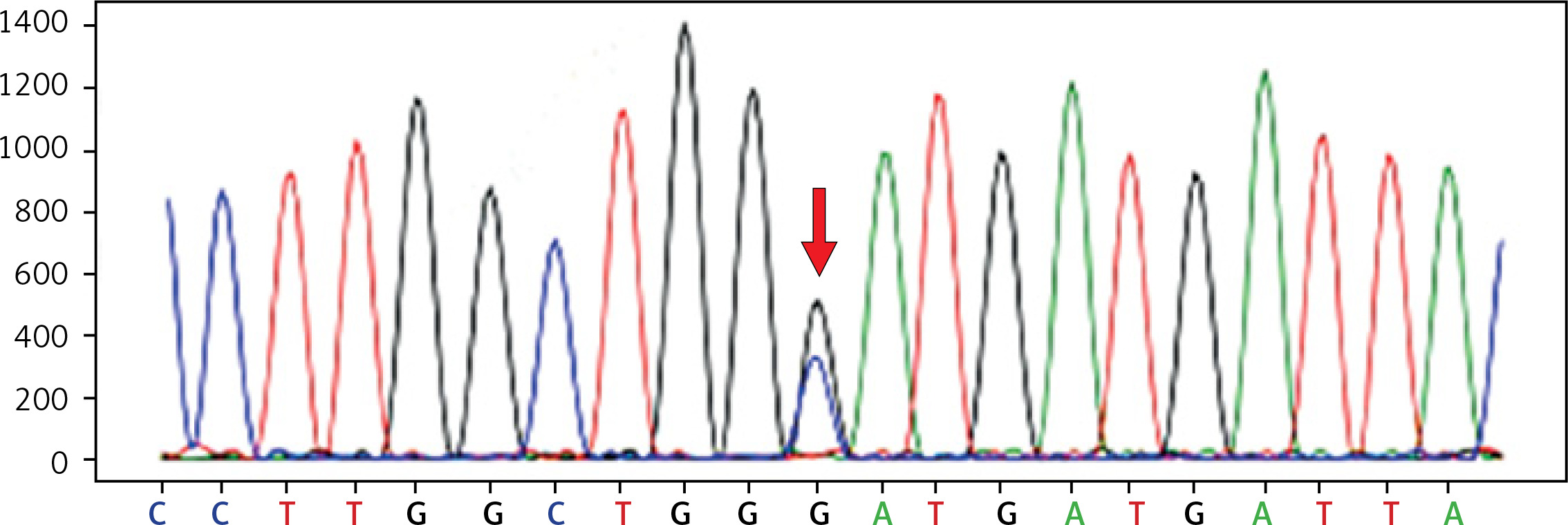
WES identified a heterozygous mutation in exon 19 (c.2245G>C) of the GSN gene in the proband (Figure 3). This mutation leads to a change in amino acid 749, where aspartate (Asp) is replaced by histidine (His) (p.Asp749His). This alteration likely disrupts the protein’s function, which may contribute to the observed clinical phenotype in the patient. Other rare variants in the proband are listed in Supplementary Table SI.
Based on the clinical presentation, auxiliary examinations, and genetic analysis, the patient was ultimately diagnosed with GSN gene mutation-induced FAF associated with nephropathy and thrombotic microangiopathy. Initially, immune modulation therapy was recommended. The patient received treatment with plasma exchange, glucocorticoids, and supportive care. Following this treatment, the patient’s hematological parameters significantly improved, and her neurological symptoms resolved. After 2 weeks of hospitalization, she was discharged in stable condition and continues to be monitored as an outpatient for ongoing management.
HGA is an autosomal dominant hereditary disease, and clinical presentation typically does not include renal amyloidosis. Patients homozygous for GSN mutations tend to exhibit severe nephrotic range proteinuria in the later stages of the disease, leading to end-stage renal failure. Renal failure is rare in the heterozygous form [4]. In our case, the patient initially presented with nephrotic proteinuria, and kidney pathology revealed focal segmental glomerulosclerosis-like lesions (FSGS). This atypical presentation for HGA is notable as renal involvement is seldom the initial manifestation. Nikoskinen et al. [5] described renal involvement in only 3% of their cohort of 227 Finnish patients with gelsolin amyloidosis, highlighting the rarity of such presentations.
The kidney lesions in our patient improved after hormonal and immunosuppressive therapy, which aligns with the treatment approach for FSGS. However, the subsequent development of thrombotic microangiopathy (TMA) is a novel finding in the context of HGA. TMA is characterized by microangiopathic hemolytic anemia, thrombocytopenia, kidney injury, and, in our patient, acute cerebral infarction [6]. We ruled out secondary causes of TMA, including infections, drugs, malignant hypertension, and autoimmune diseases. Additionally, genetic sequencing results excluded hereditary forms of TMA, such as complement-associated HUS, cobalamin defects, and mutations in the diacylglycerol kinase epsilon gene. The pathophysiology of TMA involves vascular endothelial injury caused by various factors, leading to the exposure of the subendothelial layer and subsequent thrombosis [7, 8].
The presence of nephropathy and thrombotic microangiopathy in our patient suggests that there may be a differential response to mutations in the GSN gene. The mutation identified in this study (c.2245G>C, p.Asp749His) is novel and has not been previously reported. Located in exon 19, this mutation differs from the more common GSN mutations (G654A or G654T) typically found in exon 4 [9]. Recent studies have shown that gelsolin is closely related to thrombosis [10, 11]. Gelsolin plays a crucial role in actin filament regulation, which is vital for platelet function and thrombus formation. The identified mutation may alter gelsolin’s function, predisposing the patient to both amyloidosis and thrombotic complications. This case underscores the importance of considering gelsolin amyloidosis in young patients presenting with proteinuria, even without typical FAF clinical features. Furthermore, it expands the phenotypic spectrum of GSN mutations to include thrombotic microangiopathy, a previously unreported complication.
In conclusion, we report a novel heterozygous GSN mutation associated with an atypical presentation of gelsolin amyloidosis, featuring early-onset nephropathy and thrombotic microangiopathy. This case highlights the need for genetic testing in unexplained proteinuria cases and contributes to the evolving understanding of the clinical spectrum of gelsolin amyloidosis. Further functional studies are necessary to elucidate the precise mechanisms by which this novel mutation contributes to the observed clinical phenotype.