Congenital tufting enteropathy (CTE), also known as intestinal epithelial dysplasia, is a rare congenital chronic diarrheal disease. The clinical symptoms are characterized by lifelong diarrhea, severe malnutrition and growth failure. Some patients had complications such as ophthalmology signs (photophobia, cataract, corneal erosion), atresia (rear nostril atresia, anus atresia) and other multiple deformities, which were associated with SPINT2 mutations [1]. It is a rare disorder with an estimated prevalence of 1 in 50,000 to 1 in 100,000 live births in western Europe, but appears to be higher in areas with consanguineous families of Arabic origin. The characteristic histological features of CTE include total or partial villus atrophy, non-inflammatory crypt hyperplasia and focal epithelial tufts composed of enterocytes. Currently, CTE treatment strategies mainly focus on nutritional support. Children with CTE have to rely on parenteral nutrition (PN) and eventually intestinal transplantation. In addition to Pathak et al. [2] who exposed epithelial cell adhesion molecule (EPCAM) as a gene for CTE, lots of mutations of EPCAM have also been observed. In this case, our aim was to further report a patient with chronic diarrhea and severe malnutrition by compound heterozygous mutations of the EPCAM gene.
A 6-year- and 11-month-old girl was admitted to our hospital with intermittent diarrhea for over 6 years.
The patient exhibited recurrent emesis and abdominal distension in 10 days after birth. Her vomiting was non-bloody, non-bilious and frequent. Severe watery diarrhea was noted at the age of 7 months when feeding with complete nutritional formula powder diet. The episodes of stool were about four to ten times a day. When she lived with millet porridge and rice paste instead of complete nutritional formula powder at about 2 years old, the character and frequency of stool improved. She required multiple hospitalizations after birth because of severe malnutrition and growth failure. Her vomiting and abdominal distension improved but diarrhea persisted.
She was born to a G1P1 mother at 40 + 4 weeks gestation, with a birth weight of 3100 g and height of 49 cm. Her Apgar scores were normal. Her parents are Chinese and are healthy without consanguineous mating. There were no similar diseases occurred in her family history. She could raise her head at 3 months, turn over at 6 months, sit by herself at 8 months, but to date she cannot walk by herself. She can say a series of simple words, but some words are unclear. Moreover, she was initially breastfed until 7 months and lived with complete nutritional formula powder. Then, she lived off complete nutritional formula powder with millet porridge and rice paste at about 2 years old. The current diet is liquid food (porridge) and digestible food (steamed buns).
When she was admitted, she had severe malnutrition, while she had stable vital signs. Her body weight was 12 kg (below third percentile) and measured 98 cm (below third percentile) in height. She had minimal subcutaneous fat. Her whole mouth teeth had varying degrees of enamel defect, exposed dentin and yellowing. Lungs auscultation was normal, the rhythm and strength of the heart beat were normal. Neither abdominal tenderness nor hepatosplenomegaly was observed.
Initial laboratory studies revealed the complete blood count: a white blood cell count of 6.6 × 109/l (48.7% neutrophils), a hemoglobin level of 123 g/l, and a platelet count of 415 × 109/l. The stool examinations were negative. Liver function tests, kidney and thyroid function were normal. Inflammatory markers, blood gas and electrolytes levels, serum albumin level, serum immunoglobulin levels were all normal. Her growth factor and insulin-like growth factor levels were below normal. Autoimmune screening was negative. Blood analysis of blood tandem mass spectrometry did not indicate any metabolic disease.
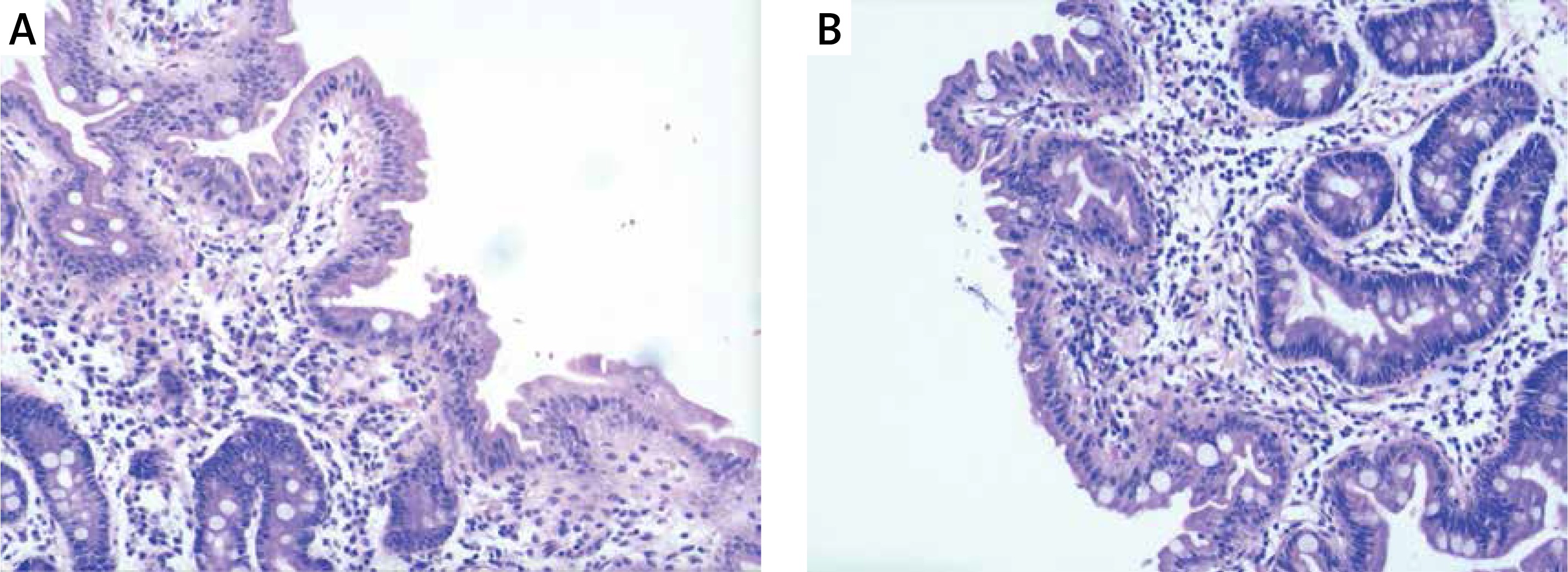
Hip X-ray showed pelvic osteoporosis. The ultrasound reads for liver, bile, pancreas, spleen, and blood vessels were normal. Histopathological findings of jejunal biopsies using light microscopy showed enterocytes with focal resembling tufts and villus atrophy when stained with hematoxylin and eosin (Figure 1). The patient was diagnosed with chronic diarrheal disease (congenital diarrhea), severe malnutrition and growth failure according to the clinical and laboratory findings. The patient was given partial parenteral nutrition during hospitalization and her diarrhea improved after treatment. In order to confirm the diagnosis, genetic testing was obtained with the consent of the parents.
Figure 1
A – Tufted enteropathy-focal epithelial clusters stained with HE 40×; B – Tufted enteropathy-villous atrophy with HE staining 40×
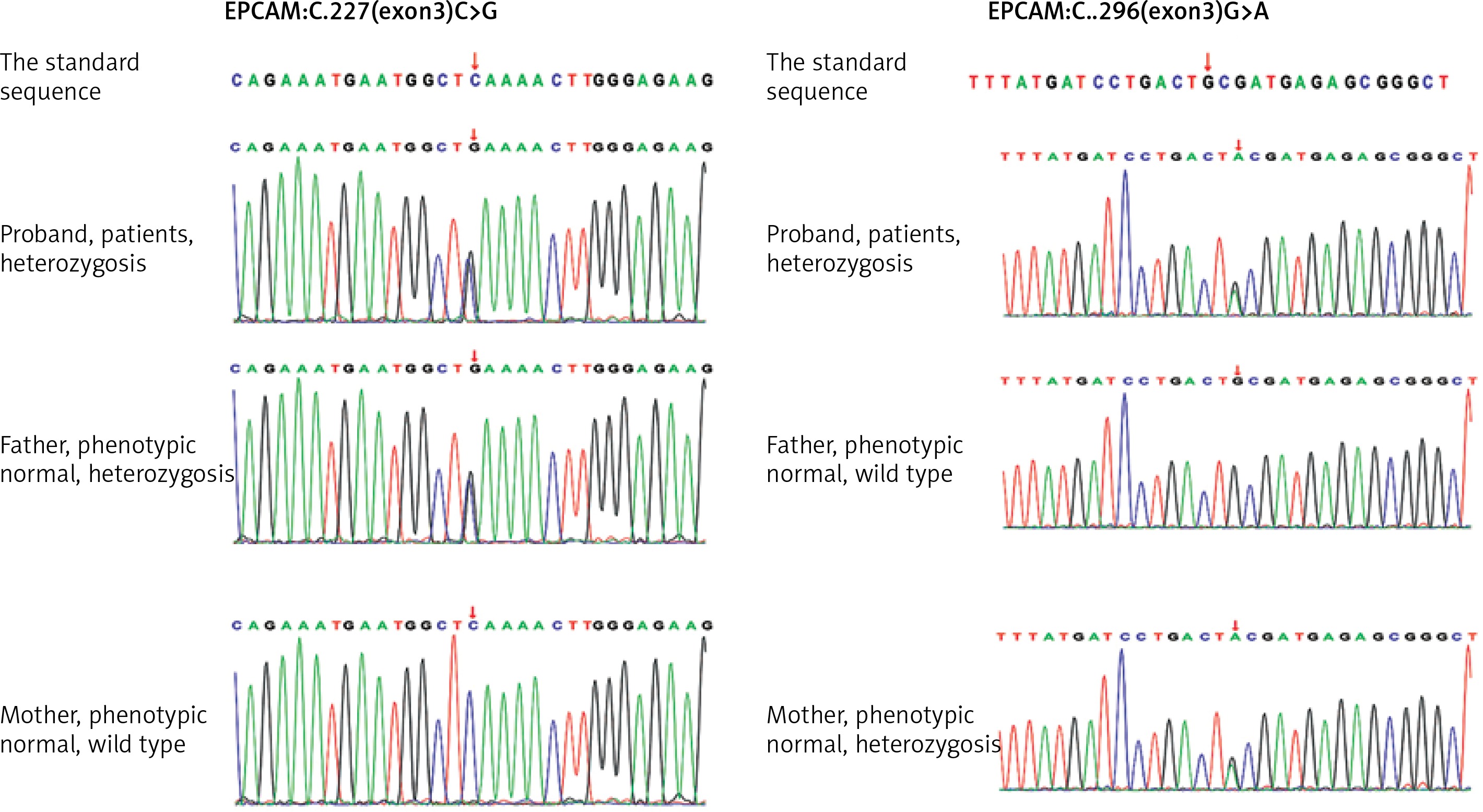
Peripheral heparin anticoagulation blood taken from the patient and her parents was sent to the Beijing Chigene Translational Medical Research Center. The peripheral blood genomic DNA was extracted using the Blood Genome Column Medium Extraction Kit (Kangweishiji, Beijing, China). The extracted DNA samples were subjected to quality controlling by utilizing Qubit 2.0 fluorimeter and electrophoresis with 0.8% agarose gel for further protocol. Protein-coding exome enrichment was performed by using xGen Exome Research Panel v1.0 (Integrated DNA Technologies, Coralville, Iowa, USA) that consisted of 429826 individually synthesized and quality-controlled probes, which targeted 39 Mb protein-coding region (19,396 genes) of the human genome and covered 51 Mb of end-to-end tiled probe space. High-throughput sequencing was performed by DNBSEQ-T7 sequencing platform, and not less than 99% of the target sequence were sequenced. The result was compared with the standard sequence. Whole-exome sequencing showed compound heterozygous mutations (c.227C>G, c.296G>A) in the EPCAM gene, among which c.227C>G is a reported pathogenic gene, and c.296G>A is a new mutant gene of EPCAM (Figure 2).
Finally, the patient was diagnosed as CTE according to her clinical manifestations, auxiliary examinations and genetic reports.
The patient received PN with electrolyte and trace elements through peripherally inserted central venous catheters. After 1 month, the patient gained up to 1 kg in weight (total body weight was 13 kg) and her diarrhea improved. After leaving hospital, she had normal diet (noodles, vegetables) and defecated 1–2 times a day with a shaped stool, her weight quickly dropped to 12.2 kg in less than a month. Then, she had to return to the hospital and received PN treatment. At the time of this writing, she was taking small amounts of oral feeds in combination with total parenteral nutrition. Diarrhea is still present but mild. Her parents rejected small bowel transplantation.
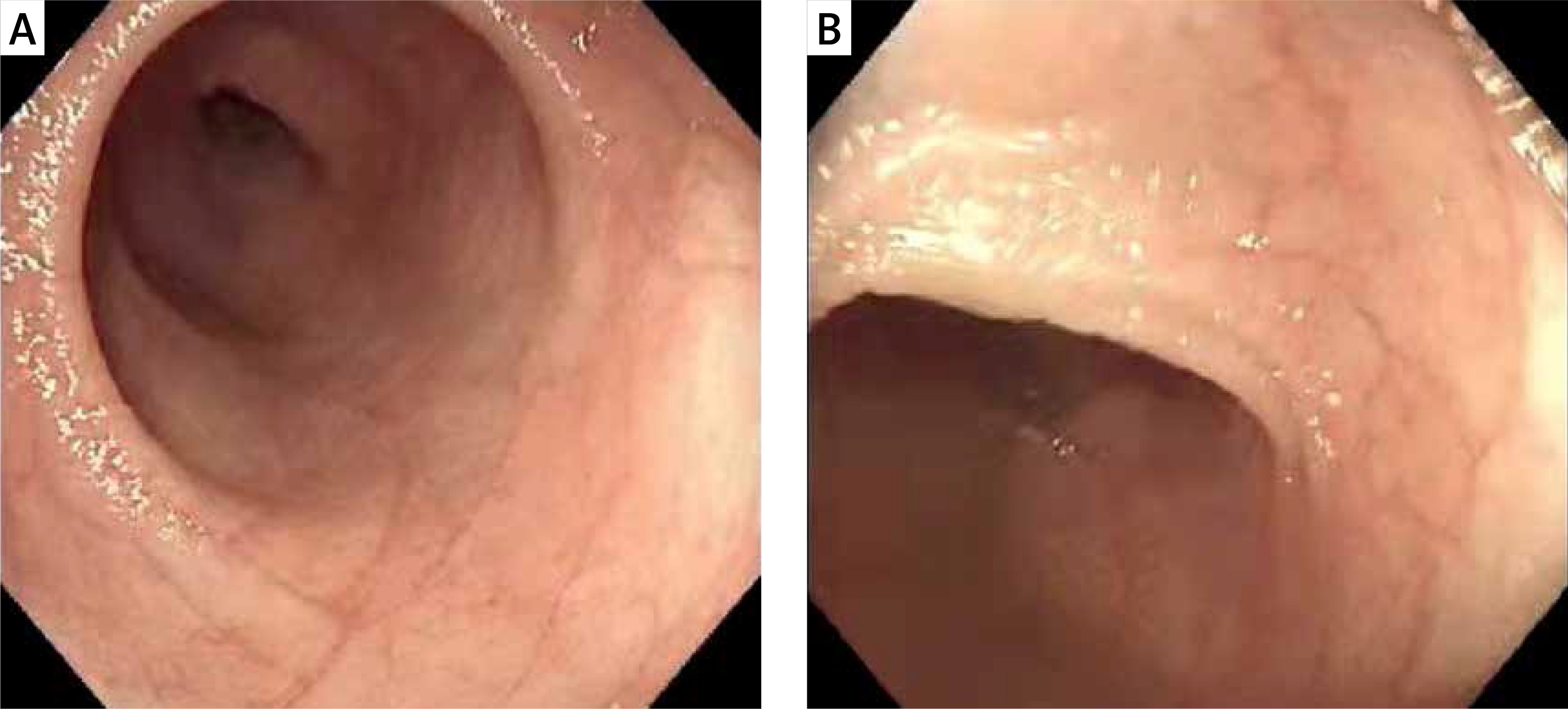
The patient in this study developed vomiting and bloating shortly after birth, followed by intractable diarrhea. There was an obvious weight loss and growth failure. Gastroscopy and colonoscopy pathological biopsy reported the upper jejunum and the terminal ileum partial villous atrophy (Figure 3). The histopathological findings of our patient were typical, but these features are nonspecific and can be seen in other enteropathies. Genetic studies revealed compound heterozygous mutations (c.227 C>G, c.296 G>A) in the EPCAM gene, which was inherited from the father and mother, respectively. The patient’s clinical manifestation and auxiliary examination accord with the CTE diagnosis.
The patient did not develop diarrhea within days after birth. However, she exhibited intractable diarrhea after changing complete nutritional formula powder diet at 7 months old. It was probably related to the heavily amount of vomiting and the low actual intestinal intake. Meanwhile, the character and frequency of stool improved when she lived with millet porridge and rice paste at about 2 years old. Considering that her intestines still retain certain functions. Besides vomiting, diarrhea, and dysplasia, she had no extraintestinal deformities such as corneal erosion and posterior nostril atresia. Combined with the clinical symptoms and compound heterozygous mutation of EPCAM, it was considered to be a milder isolated CTE. After receiving PN during hospitalization, her weight increased shortly and diarrhea improved. Her weight gradually decreased without PN after leaving hospital. It is considered that the child is still dependent on PN.
A total of 16 articles about CTE caused by the EPCAM gene, containing 81 cases, were retrieved. A summary of 81 patients found that the vast majority of reported EPCAM mutation-caused CTE patients were from Europe, Northwest Africa, the Mediterranean and Arab regions, while 3 patients were from China and 2 patients were from South Korea. Since then, more than 40 EPCAM mutations have been discovered. Many CTE patients (56 of 81 patients) were homozygotes and most of the remainder (21 of 88 patients) were compound heterozygotes for mutations in EPCAM, finally, there are a few gene fragments or complete gene deletions. The virulence gene for CTE in EPCAM was classified into four parts: chromosomal deletion, non-coding/splicing, frameshift/truncation, and missense mutation [3]. Only 3 kinds of EPCAM mutations have been reported in 5 or more patients: c.498insC (20 patients, 15 homozygous), c.556-14A>G (14 patients, 10 homozygous), c.491+1G >A (10 patients, 3 homozygous). Frameshift mutation (c.498insC) is the most common form of mutation in all CTE patients, c.556-14A>G is the most common intron mutation, which forms a new receptor splice site in intron 5., which affects the splicing of EPCAM transcription. All CTE patients (81 patients) showed severe persistent diarrhea in the first month after birth. The disease outcomes included: 78 patients treated with PN, 1 patient rejected PN, the treatment of 2 patients was unknown; 9 patients accepted intestinal transplantation; 8 patients (of 68 patients) died, no patients of intestinal transplantation (9 patients) died.
EPCAM (formerly known as TACSTD1 or CD326) is a conserved type I transmembrane glycoprotein of 35 kDa, consisting of an extracellular domain, a single transmembrane domain and an intracellular domain. The EPCAM gene maps to chromosome 2p21 (including 9 exons and 8 introns), and is expressed on the basolateral membrane of multiple epithelial cells and plasma cells. Mutated EPCAM causes defects in the cell barrier function and destruction of cell-to-cell connections, but not intestinal cell function defects [4]. EPCAM mainly plays an important role in maintaining intestinal epithelial homeostasis, and it participates in a variety of cell functions, including proliferation, migration, adhesion, differentiation and cell signaling.
It has been nearly 25 years since Reifen et al. [5] first discovered CTE in 1994 and over a decade since the identification of the genetic etiology of the disease. Although the exact pathogenesis of CTE remains poorly understood, it can be determined that the mutations of EPCAM and SPINT2 directly or indirectly affect the stability of EPCAM protein. Mutation of EPCAM in CTE results in protein conformation changes, which alter its stability and lead to proteolytic degradation, increased internalization and accumulation in the endoplasmic reticulum (ER). The misfolded protein does not leave the ER and rather accumulates, resulting in ER stress, which ultimately leads to the unfolded protein response (UPR). There are two broad clinical subtypes of CTE: isolated CTE and CTE associated with syndromic features. Isolated CTE manifests with intestinal symptoms such as diarrhea, vomiting, and abdominal distension. Apart from the manifestation of the primary intestinal symptoms, patients with a syndromic form of CTE develop extra-intestinal symptoms such as corneal erosion, nostril atresia, and anal atresia.
Previous diagnosis mainly relied on clinical symptoms, intestinal histopathological examination, immunohistochemistry, etc. However, clinical symptoms and pathological biopsy lack specificity. Given that both EPCAM gene and SPINT 2 gene mutations can directly or indirectly affect EPCAM protein, immunohistochemistry can only confirm EPCAM protein mutations and cannot confirm the specific gene phenotype. Therefore, when patients are suspected of having CTE, EPCAM gene detection is better than small bowel pathological biopsy and immunohistochemistry. CTE is a severe condition without curative treatment to date, its treatment is mainly nutritional support. Most CTE patients with neonatal diarrhea present total PN dependency, but some patients with a milder phenotype only require partial TPN ranging from 3 to 6 times per week [6, 7]. Only the occurrence of life-threatening PN complications should justify considering intestinal transplantation.
We herein report a novel compound-heterozygous EPCAM mutation in congenital tufting enteropathy in China, bringing the total cases in the country to four. At present, the number of EPCAM mutations and CTE patients carrying the same gene mutations is limited, which severely limits people’s research on the disease. Therefore, more genotype-phenotype correlation studies are required to guide physicians to become aware of this disease when patients present with refractory diarrhea with early disease onset.