Nasu-Hakola Disease is a rare disease, first described by Nasu et al., who reported membranous lipodystrophy with autopsy findings of peculiar polycystic lesions in the skeletal and nervous systems [1, 2]. Almost simultaneously Hakola described an unknown hereditary disease with dementia and lipomembranous polycystic osteodysplasia [2, 3].
The disease is caused by the mutation in genes TREM2 (Triggering Receptor Expressed On Myeloid Cells 2) or TYROBP (Transmembrane Immune Signalling Adaptor) and is inherited in an autosomal recessive manner [4]. Nasu-Hakola Disease can be diagnosed in patients with the characteristic clinical and radiographic features [5], which include polycystic osseous lesions, frontal lobe syndrome, and dementia [6]. Although there are multiple, more common causes of dementia [7], and the occurrence of frontal lobe syndrome features, such as agitation, euphoria, or emotional lability might be seen in patients with cognitive impairments [8], their combination with bone cysts is known to be unique for dementia in the course of Nasu-Hakola Disease. Genetic testing may be performed in the case of inconclusive findings.
A 62-year-old woman was admitted to the hospital with the occurrence of epileptic seizures to modify the antiepileptic treatment in September 2022. The diagnosis of epilepsy was made in 2016. The patient had been treated with levetiracetam. Before the hospitalization, an increased frequency of seizures had been observed. Of note, behavioural problems with aggression and agitation were observed after seizure episodes. She was also diagnosed with organic personality disorders and dissociative disorders in 2017. She had an ischaemic stroke in July 2017. Since then, she had been kept under observation due to epilepsy and progressive cognitive decline. Neuropsychological assessment was performed repeatedly each time the patient was hospitalized.
During previous hospitalizations in the Neurology Department, cerebral autosomal dominant arteriopathy with subcortical infarcts (CADASIL), progressive multifocal leukoencephalopathy (PML), metachromatic leukodystrophy, adrenoleukodystrophy, as well as infectious, paraneoplastic, and autoimmune aetiology of the presented conditions were excluded. Blood tests in 2017 suggested autoimmune hypothyroidism, and therefore hormonal substitution treatment was initiated. The ineffectiveness of this therapy with further progression of white matter lesions excluded Hashimoto encephalopathy from the differential diagnosis.
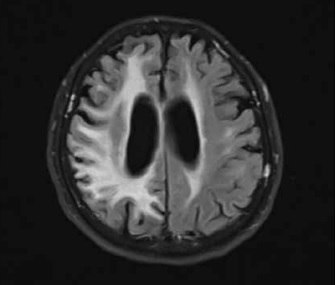
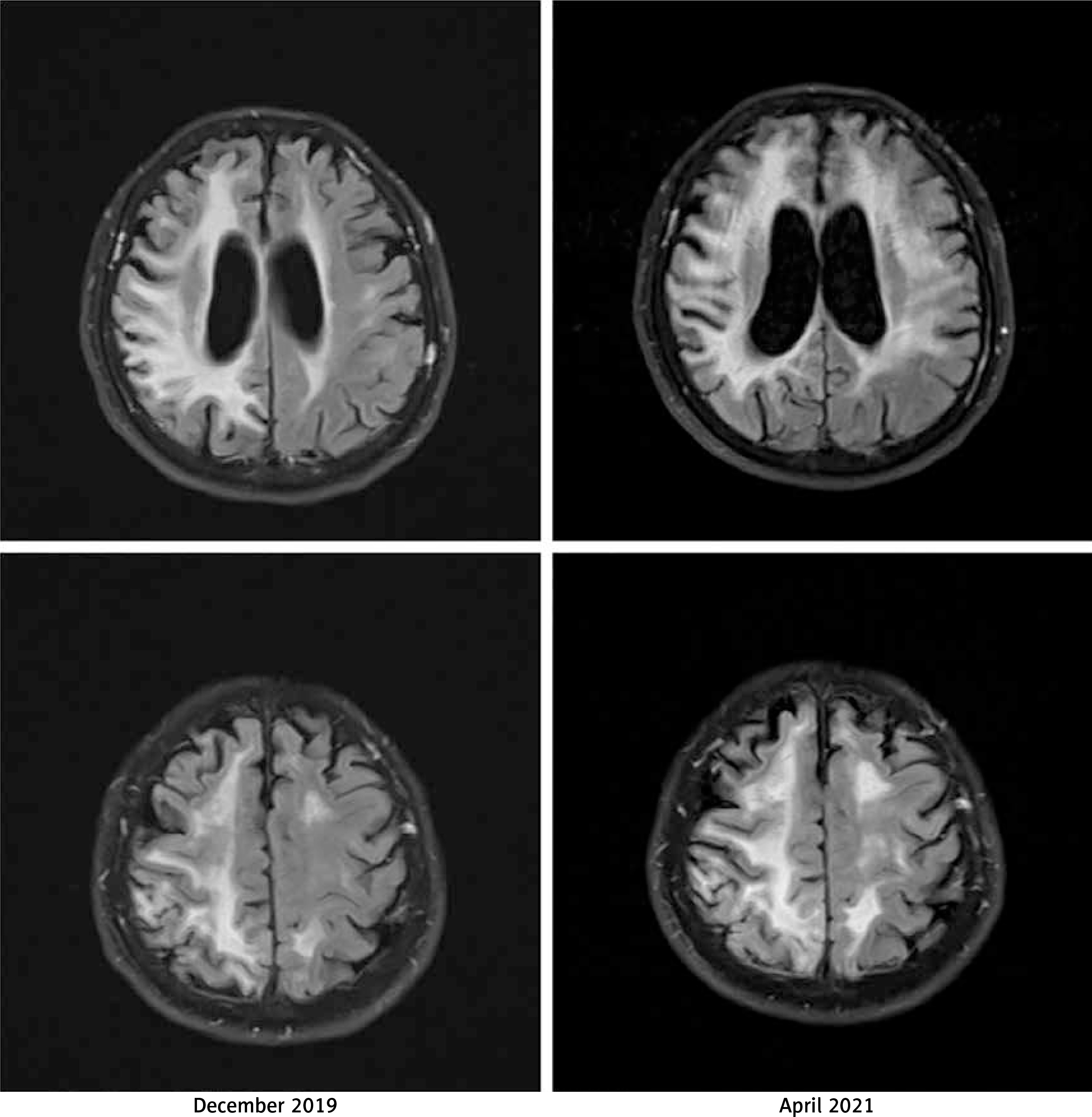
A neurological examination performed on admission in April 2021 showed left homonymous haemianopia, left-side paresis, hemiparetic gait, and hypertonia in both the upper and the lower left limbs. Signs of cerebellar damage, including dysdiadochokinesia and cerebellar dysarthria, were observed. Magnetic resonance imaging (MRI) did not show any acute pathologies. Bilateral (but more severe in the right hemisphere) diffuse cortical, subcortical, and periventricular lesions of white matter were described consistently with the previously performed imaging. Gliosis and periventricular atrophy resulted in secondary ventricular widening visible on the cross-sectional imaging showing the body of lateral ventricles in the axial plane. Moreover, the progression of the abovementioned findings was observed while comparing images from different hospitalizations. An MRI from April 2021 shows changes similar to but more severe than those seen in the examination performed in December 2019 (Figure 1). The first 2 days of hospitalization brought a clinical improvement in the Patient’s condition.
Figure 1
The comparison of non-contrast fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging (MRI) images from (i) December 2019 (left column) and (ii) April 2021 (right column). The images present a progression of cortical and subcortical diffuse lesions of white matter (more visible in the right hemisphere), periventricular atrophy, and gliosis. The image corresponds to diffuse leukoencephalopathy with secondary ventricular widening
During the same hospitalization, a neuropsychological examination focusing on cognitive-behavioural functioning was conducted. The patient presented signs of both cortical and subcortical neurocognitive disorders, especially affecting speech, attention, and visuospatial functions, and importantly, progression was observed in comparison to the previous examination. More precisely, the scanning speech pattern and paraphasia were observed. Attention disturbances manifested as extinction and difficulties in attention switching. Visuospatial functions were severely deteriorated with co-occurring left homonymous hemianopia. Additionally, moderate executive dysfunction was present. Furthermore, organic personality disorders were diagnosed along with cognitive distortions and bradyphrenia. It is important to note that symptoms observed during the neuropsychological examination, such as speech and attention dysfunctions, personality changes, and emotional instability, accompanied by behavioural disturbances appearing after epileptic seizures, are part of the range of symptoms in frontal lobe syndrome (Table I). The profile of neurocognitive disorders suggested dementia in the course of Nasu-Hakola disease.
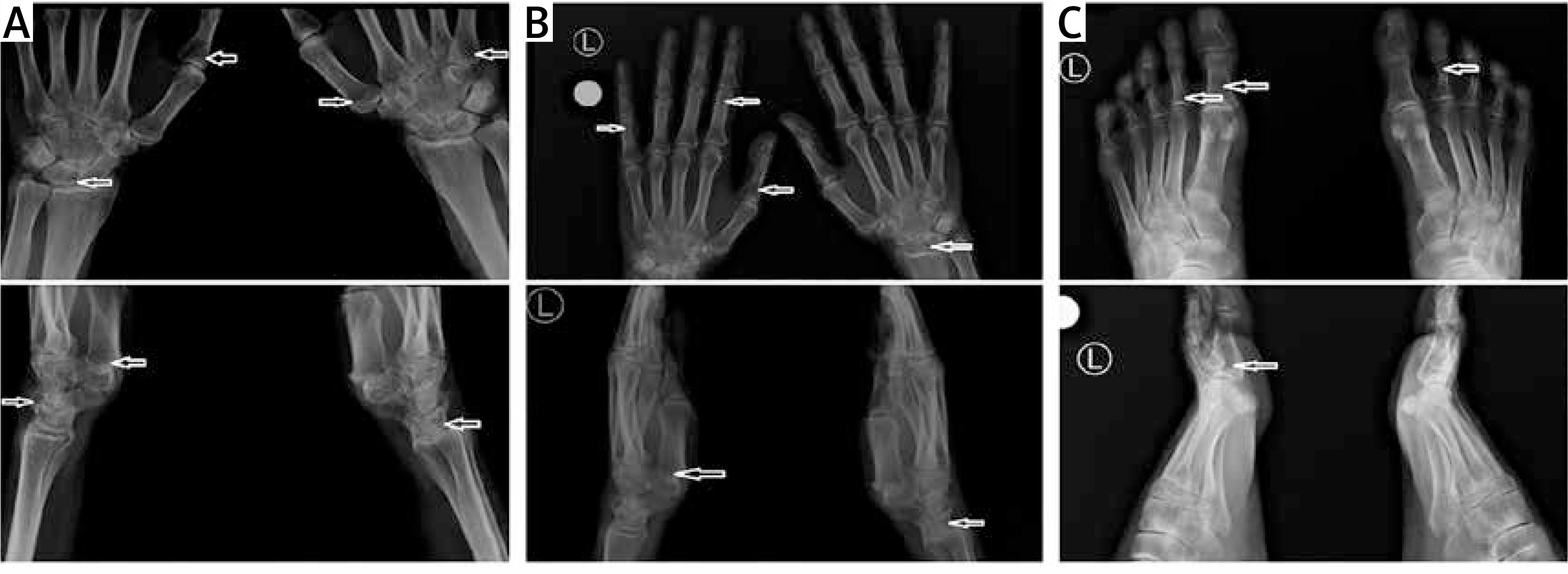
Taking into consideration the suspicion of Nasu-Hakola disease, hand and foot radiography (RG) was performed. The examination showed a single cyst within the right scaphoid and lunate bone, and minor cysts in the capitate and the hamate bones bilaterally (Figure 2 A). Additionally, the imaging revealed mild cyst lesions in the phalanges of both the right and the left hand (right: proximal phalanges of the second, third, and fourth finger; left: proximal phalanges of the third and fifth finger) (Figure 2 B). Moreover, in foot radiography, single cysts were found in the proximal phalanx of the first toe and the first metatarsal (both bilaterally) (Figure 2 C). This radiological image is highly characteristic of Nasu-Hakola disease; no other cause of dementia is known to be accompanied by polycystic bone lesions.
Figure 2
Bone cysts visible on the X-ray (arrows). The upper images are anteroposterior projections, and the lower images are lateral projections. A – Radiographs of the right and left wrist. B – Radiographs of the right and left hand. C – Radiographs of the right and left foot
The RG imaging, neurocognitive disorders, and the presence of frontal lobe syndrome suggested a Nasu-Hakola disease. It was decided not to conduct genetic testing due to certain clinical diagnosis, high costs, and lack of influence on further therapeutic management.
At the end of current hospitalization, the patient was discharged home after the modification of epilepsy treatment by the addition of valproic acid.
The described patient presented progressive dementia, skeletal cyst lesions, and some features of frontal lobe syndrome, which implied the diagnosis of Nasu-Hakola disease (Table II), also known as polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL). However, the disease is known to lead to severe cognitive impairments, a vegetative state, and death at a younger age, which were not seen in the case of described patient. Taking into consideration the occurrence of all the characteristic features of this disease as well as the late onset and gentle course of the disease, a mild type of Nasu-Hakola disease was proposed as a diagnosis. Although the presence of characteristic features crucial to the diagnosis of Nasu-Hakola disease is undeniable, there are some differences from the typical course of the disease. Firstly, the patient meets the criteria of presenile dementia, defined as the onset of cognitive disorders before the age of 65 years; however, Nasu-Hakola disease, with its typical course, usually has an onset at a significantly younger age. Furthermore, the radiologically demonstrable polycystic osseous lesions usually cause pathological fractures and disability in patients with a typical course, which was present in previously described cases but does not appear in this case. Importantly, some of the features of frontal lobe syndrome, such as emotional agitation and dysphoria, were observed to be variable in time instead of being constantly present. All the above strongly support the diagnosis of a mild type of Nasu-Hakola disease. Although only genetic testing may confirm the mutation in one of the genes responsible for the disease development, the accessibility of this diagnostic method remains limited, and the costs are usually very high. Moreover, the therapy of Nasu-Hakola disease is symptomatic, with the use of orthopaedic devices or surgery in individual cases and anti-epileptic drugs preventing epileptic seizures. No causative therapy has been developed so far; therefore, the treatment remains the same regardless of the results of genetic testing. None of the family members have presented any symptoms; therefore, they did not insist on a molecular diagnosis. Of note, something that distinguishes diseases inherited in an autosomal recessive manner is their occurrence mainly in siblings of the proband. Importantly, the described patient was an only child. Given the above, in most cases, including the presented one, genetic testing is not conducted, being reserved only for inconclusive cases. Because the described patient presented all the characteristic features of the disease (both clinical and radiological), the diagnosis of Nasu-Hakola disease is justified.
Table II
The comparison of the features in favour of the diagnosis (occur significantly more often in the typical course of Nasu-Hakola disease) and the features against the diagnosis (are less typical for the disease)
| Features for the diagnosis | Features against the diagnosis |
|---|---|
Until now, only single cases of Nasu-Hakola disease have been reported; thus, our knowledge of the aetiology and possible manifestations remains very limited. The presented case of a mild type of Nasu-Hakola disease was reported for the first time, being previously undescribed. Because genetic testing is rarely performed, the disease may be underdiagnosed, and the possible number of cases with a mild type of the disease may be higher, especially among demented patients. Importantly, the decision about the referral for an X-ray may be crucial for the diagnosis of the patient with an unusual course of dementia. Therefore, this case study aims to prompt clinicians to more thoroughly examine demented patients, with increased awareness and knowledge about the disease.